
烟熏鱼中苯并(a)芘残留物的测定
2015-10-09 05:25刘伶俐伍远安李传武李小玲陈湘艺黄向荣
湖南农业科学 2015年3期
刘伶俐,伍远安,洪 波,李传武,刘 丽,肖 维,李小玲,陈湘艺,黄向荣
(湖南省水产科学研究所,湖南 长沙410153)
苯并(a)芘简称Bap,是多环芳烃类化合物(PAHs)的代表,具有多个同分异构体,他的产生机理是由于食品中有机成分在高温缺氧条件下裂解产生碳氢自由基结合成乙炔,最终转化为苯并(a)芘[1]。现已证实为持久性难降解有机污染物,对人类健康和环境造成极大危害,被列为广泛关注的影响食品安全的物质之一。因此快速、准确地分析烟熏鱼中苯并(a)芘残留物是食品安全和保障人体健康的迫切需求。
烟熏鱼中苯并(a)芘的测定方法还没有执行标准,目前其检测方法主要为高效液相色谱法[2]、薄层层析法[3]、气相色谱串联质谱法[4],常用的前处理技术主要为薄层层析法[5]、Triton X-100 法[6]、微柱管法[7],而现有的文献及方法多存在前处理时间长,溶剂用量大,实验操作繁琐等诸多问题。由于烟熏鱼的基质成分较为复杂,样品处理稍有不当,极易造成试样中的苯并(a)芘分析损失和干扰,样品的前处理十分重要。试验尝试采用全自动凝胶净化系统净化,利用系统良好的重现性和安全性,结合超高效液相色谱仪快速、稳定的分析能力,制定一个适用于烟熏鱼中苯并(a)芘残留物的检测方法。
1 材料与方法
1.1 仪器
超高效液相色谱仪配荧光检测器:WatersACQuity,美国Waters 公司;平行蒸发器:Q-101,瑞士Buchi 公司;高速冷冻离心机:Z323K,德国Hemel 公司;超声波清洗器:KQ5200DE,昆山超声波仪器厂;全自动凝胶净化系统:Auto clean,美国Lab Tech 公司,色谱柱填 料Bi0-Beads S-X3; 氮 吹 仪:N-EVAP, 美 国Organomation Associates 公司;色谱柱:BCH C18,美国Waters 公司;微孔滤膜:0.20μm 有机滤膜。
1.2 试剂
甲基睾酮标准品:98.5%,国家标准物质研究中心;甲醇、乙腈、环己烷、石油醚、三氯甲烷:色谱纯,美国Tedia 公司;试验用水:≥18.2 MΩ/cm。
1.3 实验方法
1.3.1 标准储备液、中间液的配制 准确称取苯并芘标准品10.15 mg,加入少量乙腈溶解,转移、定容至100 m L 容量瓶中,该储备液浓度为100 μg/m L,避光储存在4℃冰箱中。
准确吸取1.00 m L 上液于100 m L 容量瓶中,乙腈定容,该中间液浓度为1.00 μg/m L,可短时间保存。
1.3.2 工作曲线溶液的配制精确吸取一定量的中间液于50 m L 容量瓶中,乙腈定容,配成质量浓度为0.000 5、0.001 0、0.005 0、0.010 0、0.020 0 μg/m L 的标准系列。
1.3.3 液相色谱分析条件 色谱柱:BEH C18(2.1 mm×100 mm,1.7 μm);流动相:乙腈+水(75%+25%);流速:0.2 m L/min;荧光检测器:激发波长362 nm,发射波长407 nm;柱温:30℃;进样量:20 μL。
1.3.4 样品提取 准确称取样品5.00 g 于50 m L 具塞离心管中,加入5 m L 乙腈、10 m L 1 mol/L KOH 溶液,于50℃的水浴中皂化1 h,皂化完毕后,冷却到室温,再加入10 m L 正己烷,涡旋震荡提取3 m in,取出水相,然后在有机相中加入5 g 无水硫酸钠充分混匀,液层转入至50 m L 鸡心瓶内,在45℃水浴中旋转蒸发,浓缩约2 m L 时取下鸡心瓶,待净化。
1.3.5 GPC 净化(样品净化) 将鸡心瓶中的液体转移至样品瓶中,使用二氯甲烷定容至10 m L,进行GPC 净化。GPC 定量环体积为5 m L,流动相使用二氯甲烷,柱流速4.5 m L/m in,收集时间6.8 ~16.0 m in。馏分收集后氮吹浓缩至干,用乙腈溶液定容至1 m L,经0.20 μm 的微孔滤膜过滤,供超高效液相色谱仪测定。
2 结果与分析
2.1 提取溶剂的选择
目前文献中用作苯并(a)芘残留物的提取剂种类繁多,利用其易溶于苯、正已烷等有机溶剂的特性,以正己烷、甲醇、乙腈为提取剂时均有一定的提取效率。此外,有研究表明苯具有更为优越的提取效率,但考虑到苯的强致癌性质,出现的文献中大都避免采用这种提取溶剂。
对于烟熏食品,苯并(a)芘不仅残存于脂肪颗粒中,还能渗透于整个坚实的肌肉组织中;处理烟熏鱼类高油脂、高物理强度样品,在采用乙腈作为提取剂,将提取出来的油脂通过KOH 进行皂化处理,既有效地除去油脂类杂质,同时不会影响残留物的提取效率。试验还采用环己烷、三氯甲烷和石油醚作为提取剂进行样品的处理,通过计算回收率比较相应的提取效率,相关数据见表1。

表1 不同提取剂的提取效率
2.2 净化条件的选择
分别采用其他文献中利用效率和作用效果最好的Folorisil SPE 小柱[8]和C18 小柱[9]作为净化手段与本试验方法中所采用的凝胶净化系统作对比试验。
弗罗里硅土是高选择性的吸附剂,在正相条件下能够从非极性基质中强烈吸附极性分析物,对多环芳烃类化合物有较强的选择吸附能力,目前在实验室应用较多;而C18 小柱是一种更为广泛使用的萃取小柱,但对苯并(a)芘残留物不具备很高的选择吸附能力和净化作用;实验室尝试使用全自动凝胶净化系统处理提取后的试样,并对这三种净化途径的样品作超高效液相色谱进样分析和重现性(n=6)测定。发现经C18 小柱处理的样品图谱基质强度相应最大,非目标组分种类较多(图1);而Folorisil 小柱选择吸附能力与全自动凝胶净化系统的相当,样品的净化效果稍差(图2),但平行试样的相对标准偏差(RSD=8.86%)明显高于后者(RSD=3.25%)。故实验采用全自动凝胶净化系统净化样品提取液(图3)。
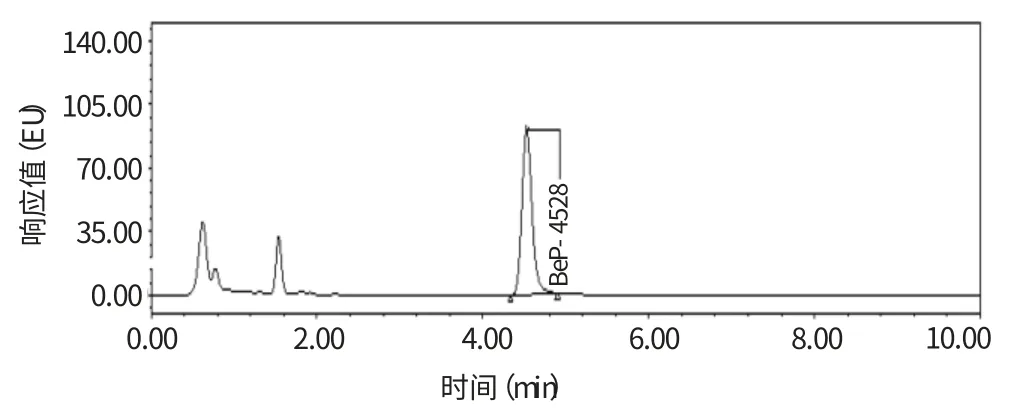
图1 全自动凝胶净化系统净化的样品图谱
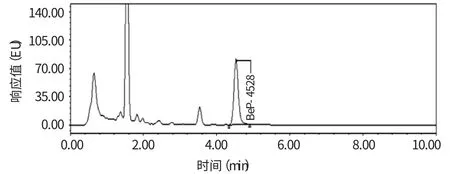
图2 Folorisil SPE 小柱净化的样品图谱

图3 C18 小柱净化的样品图谱
2.3 检测波长的确定
食品中苯并(a)芘的荧光测定方法在检测波长选择有多种报道,发射波长主要采用400~410 nm,而激发波长的选择区间较大,在290~380 nm。例如李念念等[10]采用的激发波长365 nm、检测波长410 nm,李旭华等[11]采用的激发波长296 nm、检测波长404 nm,而SC/T 3041-2008 标准方法[12]采用的激发波长297 nm、检测波长405 nm。
实验将苯并(a)芘储备液用乙腈溶液溶解后,分别从样品中测定提取其最大激发波长(Ex)和发射波长(Em)。发现该物质的最大吸收波长为407 nm,而激发波长出现了两处,分别为294.6 nm 和362.2 nm,采用Waters FLR 检测器的多通道测定功能,从两个激发波长对标液和样品采集的分析结果来看,362 nm 作为激发波长时图谱的基线平稳,出现的溶剂峰(非目标峰)种类少、相应的响应度小,所以选用362、407 nm分别作为本试验的激发波长和发射波长,具体提取图谱如图4、图5。
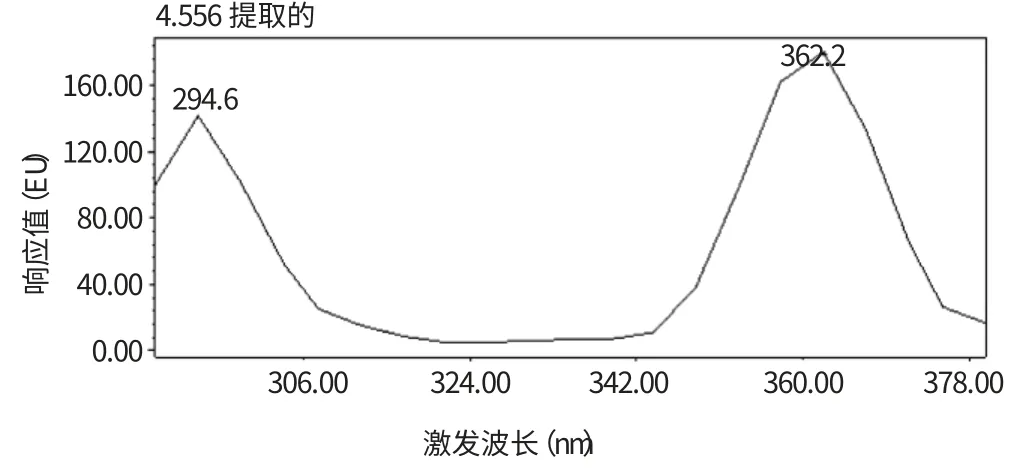
图4 最大激发波长的提取

图5 最大吸收波长的提取
2.4 实验方法评价
2.4.1 方法的线性范围 将配成浓度为0.000 5、0.001 0、0.005 0、0.010 0、0.020 0 μg/m L 的标准系列溶液,按1.3.3 的色谱条件进行UPLC 进样分析,以保留时间定性,以峰面积对质量浓度绘制工作曲线,结果表明,线性范围在0.000 5~0.020 0 μg/m L,其线性回归方程Y=579 510 X-414 899,相关系数R2=0.999 3。
2.4.2 检出限 将标准溶液逐级稀释后添加于空白样品中,按试验方法进行测定,以5 倍信噪比计算测定下限为0.5 μg/kg。
2.4.3 回收率和精密度 在最优的实验条件下,按照实验方法对翘嘴鲌、小黄鱼、麦穗鱼三类样品进行加标回收率实验,苯并(a) 芘的添加水平为1.0~50 μg/kg,每个水平作6个平行样,各添加水平的回收率和精密度如表2。结果表明,添加浓度1.0~50 μg/kg时, 回收率范围72.2%~90.4%, 相对标准偏差范围0.96%~8.26%。

表2 不同烟熏鱼苯并(a)芘回收率和精密度
2.5 实际样品的测定
应用本方法对市购的翘嘴鲌、小黄鱼、麦穗鱼等烟熏鱼进行苯并(a)芘含量的分析测定,发现一例翘嘴鲌样品中检出苯并(a)芘,含量为1.55 μg/kg,其余样品均未检出苯并(a)芘。
此外,同时采用该方法为鲜活的鲤鱼、草鱼、对虾等水产品进行苯并(a)芘含量的测定,没有出现疑似阳性样品,所采集分析的样品图谱基线平稳、基质峰少、添加回收样中目标组分无干扰、回收率高,表明该方法同样适用于鲜活水产品中苯并(a)芘残留物的测定。
3 结论
烟熏食品中基质组成颇为复杂,蛋白质种类多样,油脂含量丰富,熏制后的样品肌肉组织僵硬,给前处理带来直接影响。本实验采用最新的全自动凝胶净化系统净化处理技术进行样品的处理,与目前常用的Florisil 固相萃取柱净化、C18 小柱净化的作用效果对比,具有以下特点:①有效排除了样品中复杂的基质成分、无杂峰对目标峰的干扰,得到更为干净的色谱图背景,谱图峰形好。②该方法的回收率高,同时由于采用自动处理程序致使处理好的样品的平行性好、精密度高。③该方法前处理简便、自动化程度高,给实验室操作人员带来更多的安全保障。实验证明,该法样品预处理简单,分离效果好,明显提高了检测效率,且具备准确性和可重复性,为一种较好的测定烟熏鱼与鲜活水产品中苯并(a)芘残留物的方法,符合现代检测的发展需求。
此外,烟熏食品中多环芳烃类化合物在前处理器皿和检测设备中极易残留,建议所使用的器皿在使用前采用甲醇溶液侵泡处理,检测设备在使用前后需进行长时间的清洗。
[1]黄靖芬,李来好,陈胜军,等.烟熏食品中苯并(a)芘的产生机理及防止方法[J].现代食品科技,2007,23(7):67-70.
[2]田玉霞.反相高效液相色谱法测定植物油中苯并(a)芘含量[J].粮食与食品工业,2013,20(1):65-66、69.
[3]王焕锁,王红勇.双波长薄层扫描法测定油脂中苯并(a)芘[J].解放军预防医学杂志,1993,11(6):432-434.
[4]王春兰,汪军霞,胡 静,等.加速溶剂/固液固萃取-气相色谱/质谱法分析卷烟烟气中苯并[a]芘[J].分析化学,2013,41(7):1069-1073.
[5]天津市商检局.柱层、纸层、薄层、薄层扫描、气相和液相色谱等在测定食品中3.4苯并芘时的应用[C].天津:天津市色谱研究会,1980.123-124.
[6]杨貌端,李爱师.应用Triton X-100进行食品中苯并(a)芘的快速测定方法[J].中华预防医学杂志,1995,(7):244-245.
[7]陶顺兴,陶桂全.食品中苯并(a)芘简易快速测定方法的研究[J].食品科学,1995,16(5):48-51.
[8]吴 敏,李 政,黎翠玉,等.固相萃取-高效液相色谱-荧光检测法联合测定食品中苯并(a)芘[J].食品安全质量检测学报,2012,3(2):82-88.
[9]王红青,韩里明,屠海云,等.固相萃取-超高效液相色谱荧光法测定植物油中苯并(a)芘[J].食品科学,2012,33(14):216-218.
[10]李念念,周光宏,徐幸莲,等.高效液相色谱荧光法测定腊肉中的苯并芘残留[J].食品工业科技,2013,34(1):319-322.
[11]李旭华,段 宁,刘景洋,等.高效液相色谱法测定玉米作物中苯并(a)芘[J].环境监测管理与技术,2008,20(6):43-44.
[12]SC/T 3041-2008,水产品中苯并(a)芘的测定高效液相色谱法[S].
猜你喜欢
中国动物保健(2022年2期)2022-05-05
节能与环保(2022年3期)2022-04-26
中国中医药信息杂志(2022年4期)2022-04-26
疯狂英语·读写版(2021年5期)2021-06-15
疯狂英语·新读写(2021年5期)2021-06-04
基层中医药(2020年4期)2020-09-11
食品研究与开发(2020年9期)2020-05-08
测控技术(2018年3期)2018-11-25
食品界(2018年8期)2018-09-03
电子制作(2018年9期)2018-08-04
