
厚朴挥发性成分的HS-SPME-GC指纹图谱建立△
2015-09-25 05:54雷咪杨敏张林碧王华清刘义梅黄传奇廖朝林
中国现代中药 2015年4期
雷咪,杨敏,张林碧*,王华清,刘义梅,黄传奇,廖朝林
(1.湖北中医药大学 药学院,湖北 武汉 430065;2.湖北省农业科学院 中药材研究所,湖北 恩施 445000)
·基础研究·
厚朴挥发性成分的HS-SPME-GC指纹图谱建立△
雷咪1,杨敏1,张林碧1*,王华清1,刘义梅1,黄传奇1,廖朝林2
(1.湖北中医药大学 药学院,湖北 武汉 430065;2.湖北省农业科学院 中药材研究所,湖北 恩施 445000)
目的:应用顶空固相微萃取-气相色谱(HS-SPME-GC)技术,建立厚朴挥发性成分的指纹图谱。方法:筛选了固相微萃取(SPME)的3种不同萃取涂层的萃取头:100 μm聚二甲基硅氧烷(PDMS)、65 μm聚二甲基硅氧烷/二乙烯基苯(PDMS/DVB)和85 μm聚丙烯酸酯(PA);优化了萃取温度、萃取时间和萃取量;采用HP-50毛细管GC色谱柱,对13批厚朴药材进行比较分析,建立了厚朴挥发性成分的指纹图谱。结果:选定其中10批优质样品建立共有模式,相似度均在0.9以上,其重复性、稳定性均符合有关规定。结论:该方法简便、快速、环保,无需溶剂样品前处理,样品用量小,可作为中药材质量控制的又一手段,其试验数据为厚朴的质量控制提供了参考。
厚朴;顶空固相微萃取;气相色谱-质谱联用法;气相色谱法;指纹图谱
厚朴为木兰科植物厚朴MagnoliaofficinalisRehd.et Wils.及凹叶厚朴MagnoliaofficinalisRehd.et Wils.var.bilobaRehd.et Wils.的干燥干皮、枝皮和根皮,主产于四川、湖北[1]。现代药理研究证明,厚朴挥发油具有增加胃液、唾液分泌,加快肠胃蠕动以及健胃等药理活性[2]。
中药材挥发性成分的GC指纹图谱研究中,常采用水蒸气蒸馏法、超临界CO2流体萃取法、超声提取法提取挥发油[3-6]。水蒸气蒸馏法的高温易引起热不稳定化合物的分解[7];超临界CO2流体萃取法的设备昂贵、操作步骤繁琐,还易引入塑料增塑剂或提取出蜡质等成分[8];超声提取法的影响因素较多,药材粒度、超声波频率等设置不当会导致提取效果差异较大[9]。固相微萃取(SPME)是集萃取、浓缩、进样于一体,无溶剂样品前处理的新技术,适合对挥发性和半挥发性有机物的分析[10]。目前已有大量运用顶空固相微萃取(HS-SPME)技术分析鉴定中药材挥发性成分的报道[11-14],可见采用顶空固相微萃取气质联用法可为中药材指纹图谱的建立提供有效的方法。本研究采用HS-SPME-GC技术,对13批厚朴药材的挥发性成分进行比较分析,建立了厚朴挥发性成分的指纹图谱,为厚朴的内在质量控制提供参考。
1 仪器与材料
Agilent 6890N气相色谱仪、Chemstation色谱工作站、Agilent 6890/5973气质联用仪、Nist谱库(美国Agilent公司);BP-211D 型十万分之一分析天平(德国Satorius);手动SPME装置、100 μm聚二甲基硅氧烷(PDMS)、65 μm聚二甲基硅氧烷/二乙烯基苯(PDMS/DVB)、85 μm聚丙烯酸酯(PA)萃取头(美国Supelco公司);PC-220温度调节式磁力搅拌器(美国Corning公司);BP-211D电子天平(北京Sartorius公司)。
“中药色谱指纹图谱相似度评价系统2004版”软件(国家药典委员会)。
13批厚朴药材直接从产地采集或购于各地药店,经湖北中医药大学张林碧教授鉴定,2个批次为凹叶厚朴,其他均为厚朴。药材来源见表1。

表1 13批厚朴样品情况
2 方法与结果
2.1方法
2.1.1 色谱条件 色谱柱:HP-50毛细管柱(30 m×0.32 mm×0.25 μm);载气:高纯He;恒流:1.3 mL·min-1;程序升温:50 ℃(1 min),以4 ℃·min-1上升到140 ℃(2 min),以2 ℃·min-1上升到180 ℃,以12 ℃·min-1上升到280 ℃,恒温5 min;进样口温度:260 ℃;分流比为15∶1;氢火焰离子化检测器(FID)温度:280 ℃;尾吹气(高纯氮):35 mL·min-1。
2.1.2 质谱条件 倍增器电压为1800 V;接口温度为280 ℃,离子源温度为230 ℃;电离方式为EI,电子能量为70 eV;扫描质量范围:35~550 amu。
2.1.3 HS-SPME萃取方法 精密称取一定质量的厚朴药材粉末(过60目筛),于12 mL顶空瓶中加盖密封,插入SPME萃取头,在一定温度下,顶空萃取一定时间后拔出,立刻插入GC进样口,260 ℃解析3 min,按2.1.1项下色谱条件进行GC分析。
2.2 萃取条件的优化
——进一步增强医疗救助托底保障能力,确保年度救助限额内农村贫困人口政策范围内个人自付住院医疗费用救助比例不低于70%,对特殊困难的进一步加大倾斜救助力度。
2.2.1 萃取头的选择 分别选择PDMS/DVB(65 μm)、PDMS(100 μm)和PA(85 μm)3种不同涂层的萃取头,按2.1.1项下色谱条件、2.1.3项下方法进行GC分析,结果见图1。气相色谱图显示:PA萃取的峰个数太少;不论是出峰数目还是总峰面积,PDMS/DVB的萃取效率都优于PDMS。综合考虑以上因素,选择PDMS/DVB萃取头。
2.2.2 萃取温度的选择 加热温度分别选择50、60、70、80、90 ℃,按2.1.3项下方法操作,见图2~3。结果表明,随着萃取温度的升高,保留时间在30 min后各色谱峰峰面积明显增大,但30 min前的各色谱峰峰面积则有所减少。在90 ℃时虽然总峰面积与峰个数有所增加,但差异性明显加大,导致部分低沸点物质的响应值被淹没。综合考虑总色谱峰峰面积及峰个数,选定萃取温度为70 ℃,此时的峰面积相对较大且稳定,峰个数较多。
2.2.3 萃取时间的选择 萃取时间分别选择30、45、60、75、90 min,按2.1.3项下操作,结果见图4~5。在60 min时,萃取的总峰面积及峰个数均较高,而90 min时峰型较差,所以选定萃取时间为60 min。
2.2.4 萃取量的选择 分别选择0.05、0.2、0.4、0.6、0.8、1.0 g的厚朴细粉,按2.1.3项下操作,结果见图6~7。结果表明,萃取量太低时,峰个数及峰面积都不佳,适当增加萃取量后二者均有所提高,量太大导致峰型变差、峰面积有所下降。0.2 g时出峰个数最多,因此选择0.2 g为萃取量。
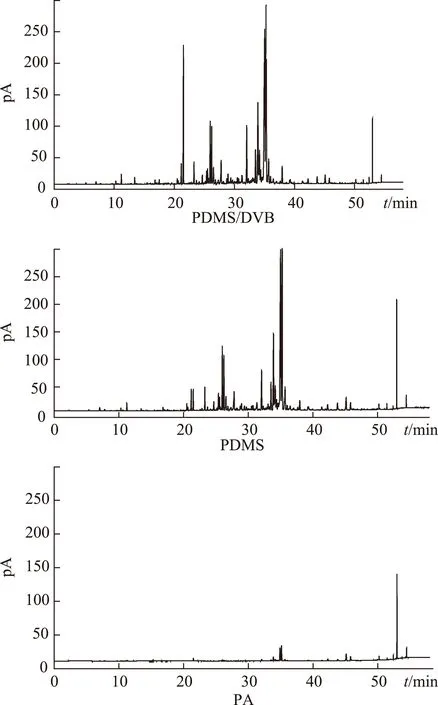
图1 3种SPME萃取头的GC色谱图
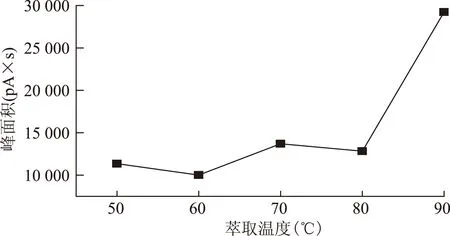
图2 萃取温度对峰面积的影响
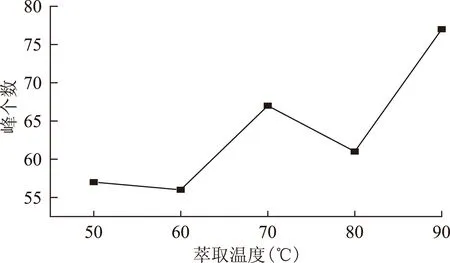
图3 萃取温度对峰个数的影响
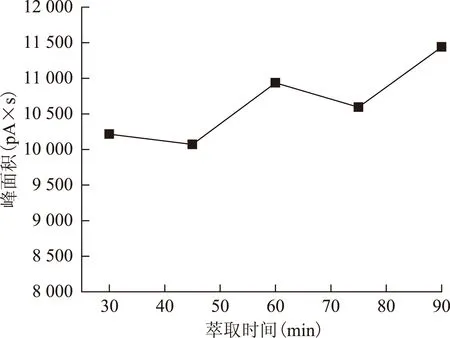
图4 萃取时间对峰面积的影响
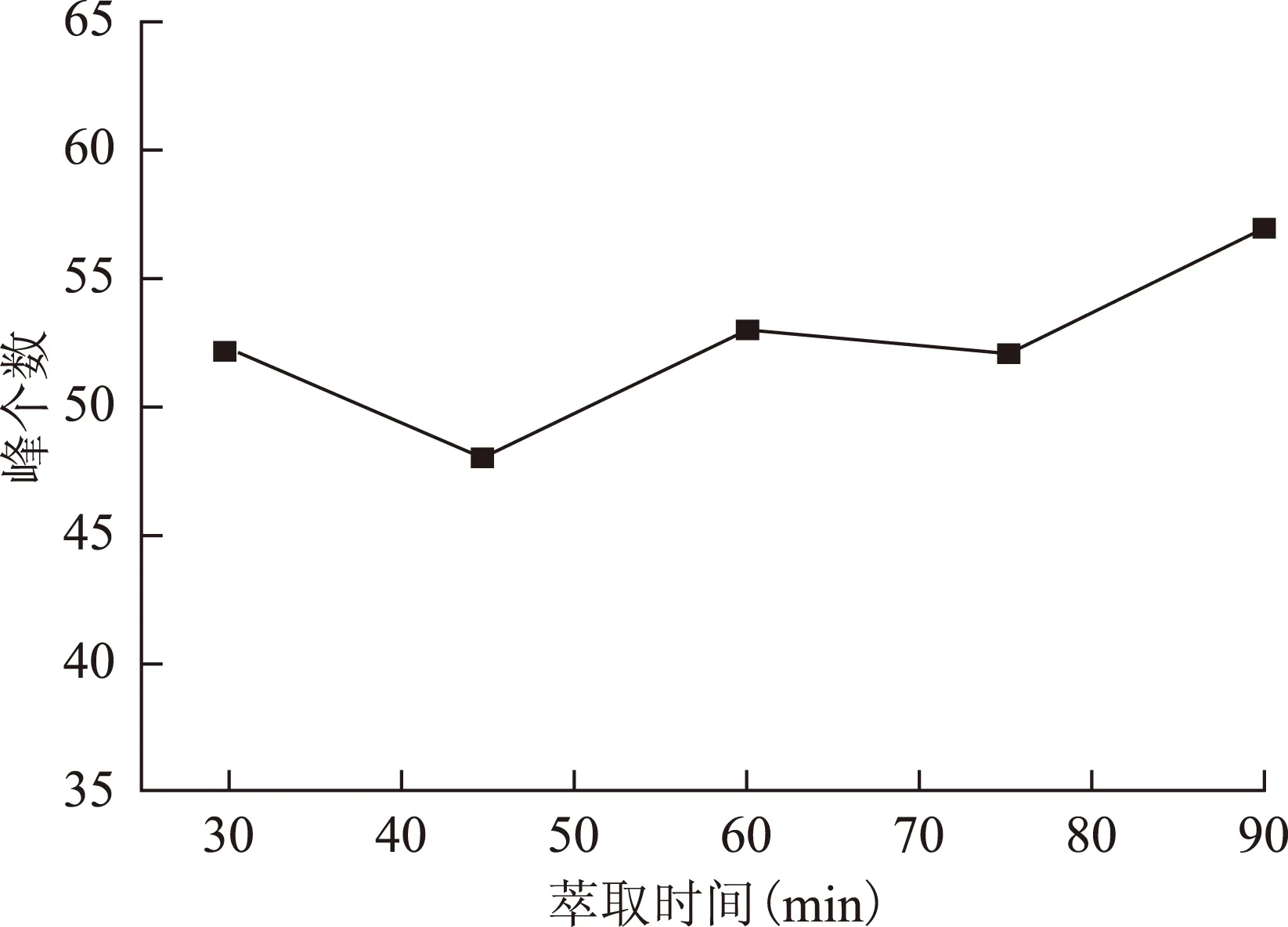
图5 萃取时间对峰个数的影响
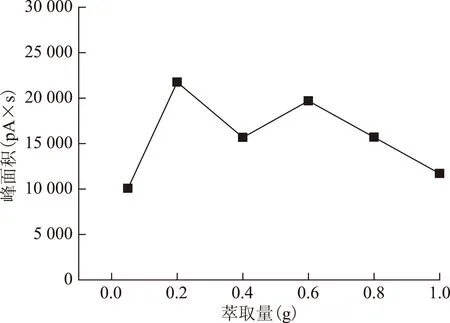
图6 取样量对峰面积的影响
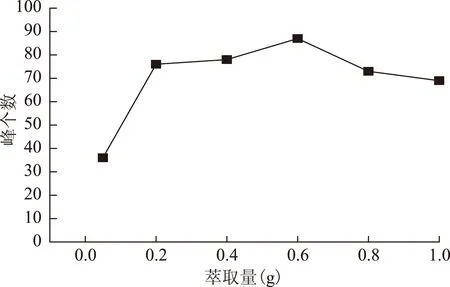
图7 取样量对峰个数的影响
2.3 方法学考察
2.3.1 重复性试验 取同一批厚朴药材(13号),按2.1.3项下操作,连续进样5次,各图谱的相似度均在0.997以上,表明该方法的重复性良好。
2.3.2 稳定性试验 取同一批厚朴药材(13号),按2.1.3项下操作,分别在0、4、8、16、24、48 h进样分析,各图谱的相似度均在0.994以上,表明样品成分在48 h内稳定。
2.4 指纹图谱的建立和分析

表2 13批厚朴样品中42个共有峰信息表

表2(续)

表3 挥发油成分GC-MS结果表
2.4.2 系统聚类分析 计算13批不同产地厚朴样品的指纹图谱中各共有峰相当于内参照峰的峰面积比值,得到42×13阶的数据矩阵,应用“SPSS”软件对其进行系统聚类分析,采用Ward法并利用欧氏距离平方(Squared Euclidean Distance)作为样品测度。聚类分析将13批厚朴样品分为2类,其中样品1、2、3、4、5、7、13归为Ⅰ类,其他归为Ⅱ类。
2.4.3 相似度评价 将聚类分析结果的第Ⅰ类和Ⅱ类中的1、2、3、5、6、7、8、9、12、13号共10批样品建立共有模式,将其导入国家药典委员会的“中药色谱指纹图谱相似度评价系统2004版A”软件,生成对照图谱,建立共有模式。生成的10批样品和对照图谱见图8。再将对照谱图和13批样品色谱图全谱导入“中药色谱指纹图谱相似度评价系统检验版(2004B)”,样品与对照图谱的相似度数据见表4。

A.参与共有模式建立的l0批样品色谱图;B.生成的GC对照色谱图1.邻聚伞花素;2.对烯丙基苯酚;3.β-芹子烯;4.α-白菖烯;5-石竹烯氧化物;6.古巴烯;8.白菖烯;9.沉香螺贴醇;12.α-桉叶油醇;13.β-桉叶油醇;14.喇叭茶醇;16.杜松烯;19.对叔丁基苯甲醚;20.十六酸;21.亚麻油酸。图8 参与共有模式建立的10批样品色谱图和生成的GC对照色谱图

样品编号相似度样品编号相似度样品编号相似度10.99760.979110.98820.98870.993120.98230.99080.997130.97040.99790.98650.993100.986
3 讨论
3.1通过SPME的3种不同萃取头选择试验可见:萃取头对挥发性成分的吸附差异性取决于其涂层材料,PDMS涂层呈非极性、PA涂层呈极性、而PDMS/DVB涂层呈双极性,故后者更适合含多种挥发性成分中药材的吸附和分析。而涂层的厚度对待测物的吸附量和平衡时间也有一定的影响。
3.2通过萃取温度、萃取时间和萃取量的优化可知:萃取温度增高能增加顶空挥发性物质的浓度,有利于被测组分的萃取。但SPME是一个放热过程,当温度高到一定程度后,化合物的萃取量反而减少,考虑到温度太高某些组分重叠更为严重,不利于样品的定性分析,所以选择了总峰面积相对较大且稳定,峰个数较多的70 ℃为试验的最佳温度。萃取时间的长短对低、高沸点物质的萃取量有显著的影响,因此对该因素进行考察是必要的。萃取量的不同,对峰面积和个数均有影响,萃取量太低时峰面积和个数都不佳;萃取量适当增加后,二者均有所提高;萃取量太大又导致峰型变差、峰面积有所下降。本试验还考察了样品粒度、进样口的解析温度等因素,最终确定了样品制备和GC分析的条件。
3.3本试验还分别考察了OV-1、HP-5、HP-50和DB-WAX柱的分离效果,结果表明OV-1柱对低沸点物质的响应较低;HP-5柱对12、13号色谱峰的分离效果不佳;DB-WAX柱的最高耐受温度为250 ℃,在程序升温至后半段时基线出现明显的漂移,故最终选择分离效果理想、基线平稳的HP-50柱。
3.4建立共有模式需要不少于10批有代表性且经专家鉴定的药材样品,本次试验数据显示13批样品的相似度均在0.9以上,未发现离群样本。可考虑增大样本批次,进行系统分析。
3.5厚朴中挥发油主要含醇、酮、酯及烯等成分,其中以桉叶油及其异构体为主。用GC-MS对部分成分进行了鉴定,根据样品的谱库检索结果和质谱图分析,α-桉叶油醇、β-桉叶油醇是厚朴的主要挥发性成分,邻聚伞花素、杜松烯、β-芹子烯、杜松烯也是厚朴的共有成分。根据文献报道和结构分析,成分中对烯丙基苯酚可能是厚朴的主要成分厚朴酚的降解产物。
[1] 国家药典委员会.中华人民共和国药典:一部[S].北京:中国医药科技出版社,2010:235.
[2] 张淑洁,钟凌云.厚朴化学成分及其现代药理研究进展[J].中药材,2013,36(5):838-843.
[3] 童巧珍,周日宝,杜方麓,等.金银花中挥发油提取工艺探讨[J].湖南中医学院学报,2002,22(12):24-25.
[4] 林琳,蒋合众,罗丽勤,等.薤白挥发油成分的超临界CO2萃取及GC-MS分析[J].分析试验室,2008,27(1):115-118.
[5] 李平,何文妮,孙博航,等.厚朴超临界提取物的化学成分研究[J].中国现代中药,2008,10(2):27-28.
[6] 付玉杰,张乃静,王黎丽,等.超声提取文冠果种仁油及GC-MS成分分析[J].植物研究,2007,27(5):622-625.
[7] 吴惠勤,黄晓兰,林晓珊,等.白豆蔻挥发油GC-MS指纹图谱研究[J].中药材,2006,29(8):788-792.
[8] 王婷婷,李清,宋爱华,等.白芷挥发油GC指纹图谱研究[J].药物分析杂志,2007,27(9):1340-1343.
[9] 李攻科.样品前处理仪器与装置[M].北京:化学工业出版社,2007.
[10] 王华清,张林碧,杨敏,等.湖北恩施厚朴顶空固相微萃取方法考察[J].医药导报,2010,29(11):1428-1430.
[11] 张家骊,王利平,袁身淑,等.醒脑静注射液指纹图谱的顶空固相微萃取气质联用研究[J].中国中药杂志,2004,29(5):466-467.
[12] 熊梅,张正方,唐军,等.HS-SPME-GC-MS法分析肉桂子挥发性化学成分[J].调味品,2013,38(1):88-91.
[13] 孙强,李梅青,吴悠.两种凤丹鲜花挥发性成分的HS-SPME-GC-MS分析[J].光谱实验室,2013,30(1):145-150.
[14] 梁欢,卢金清,戴艺,等.HS-SPME-GC-MS结合化学计量法对不同产地艾叶药材挥发性成分的比较分析[J].中国实验方剂学杂志,2014,20(9):85-90.
EstablishmentofFingerprintofVolatileCompoundsinMagnoliaeofficinalisCortexbyHS-SPME-GC
LEIMi1,YANGMin1,ZHANGLinbi1*,WANGHuaqing,LIUYimei,HUANGChuanqi,LIAOChaolin2
(1.CollegeofPharmacy,HubeiUniversityofChineseMedicine,Wuhan430065,China;2.InstituteofChineseHebalMedicines,HubeiAcademyofAgriculturalScience,Enshi445000,China)
Objective:To establish the fingerprint of the volatile compounds in Magnoliae officinalis Cortex by headspace solid phase micro-extraction-gas chromatography(HS-SPME-GC).Methods:The different three SPME fibers were screened and the conditions including extraction temperature,extraction time,sample quantity were optimized through the optimization of GC conditions.13 batches of samples were analyzed and compared using HP-50 capillary gas chromatographic column,the volatile components were identified by gas chromatography-mass spectrometry(GC-MS).Result:The samples were classified as 2 grades by cluster analysis and 10 superior samples were selected to establish the mutual models.The similarities were all above 0.9.The repeatability and stability of the method met the requirment.Conclusion:The method is simple,fast,envirnment-friendliness,less consumption of sample and can be used for the quality control.It is also reliable to evaluate the quality of Magnoliae officinalis Cortex.
Magnoliae officinalis Cortex;HS-SPME;GC-MS;GC;fingerprint
2014-08-28)
全国第四次中药资源普查(15801448584);湖北省卫生厅科研基金资助重点项目(2012Z-Y27)
*
张林碧,教授,研究方向:中药资源与品质研究;E mail:diqing2001putin@foxmail.com
10.13313/j.issn.1673-4890.2015.4.005
猜你喜欢
分子催化(2022年1期)2022-11-02
小学生学习指导(低年级)(2021年9期)2021-10-14
小哥白尼(趣味科学)(2021年11期)2021-02-28
小天使·一年级语数英综合(2020年10期)2020-12-16
环境影响评价(2020年5期)2020-12-02
皮革制作与环保科技(2020年14期)2020-03-17
中学生数理化·七年级数学人教版(2019年10期)2019-11-25
小学生学习指导(低年级)(2019年9期)2019-09-25
小学生学习指导(低年级)(2018年9期)2018-09-26
儿童时代·快乐苗苗(2016年2期)2016-10-22
