
CX26第86位氨基酸多态性与遗传性聋相关性分析
2015-09-18 06:24:32时晰董燕粉邱士伟庄伟乔月华徐州医学院听力中心徐州医学院附属医院听力中心
中华耳科学杂志 2015年4期
时晰 董燕粉 邱士伟 庄伟 乔月华徐州医学院听力中心,徐州医学院附属医院听力中心
·基础研究·
CX26第86位氨基酸多态性与遗传性聋相关性分析
时晰1董燕粉1邱士伟1庄伟1乔月华2
1徐州医学院听力中心,
2徐州医学院附属医院听力中心
目的 本研究在细胞生物学水平,对第86位氨基酸分别为Thr,Ser,Arg的CX26蛋白的膜定位生物学功能进行鉴定,进而结合临床数据分析该位点不同多态性突变的潜在致聋可能性。方法本研究借助GFP融合型慢病毒表达系统,分别将第86位氨基酸为Thr,Ser,Arg的CX26-GFP融合蛋白在小鼠螺旋神经节细胞中进行表达,并在荧光显微镜下观察融合蛋白的细胞膜定位现象。结果第86位氨基酸发生Ser突变后,CX26蛋白仍能够正常定位于细胞膜表面,并形成间隙链接结构,这一表型与(第86位氨基酸为Thr的)野生型CX26蛋白并无明显差异;只有当第86位氨基酸突变为Arg时,导致CX26蛋白功能丧失。结论这些现象表明CX26 T86R可能为潜在致聋突变,而CX26 T86S为不影响蛋白功能的氨基酸多态性突变。该结论与临床筛查经验性结果一致。
遗传性耳聋;CX26蛋白;螺旋神经节
先天性耳聋是人类常见出生缺陷疾病,发病率约为1/700-1000[1],其中一半以上为遗传性听功能障碍[2]。另有研究提出,近36.9%的儿童感音神经性聋与GJB2基因突变有关,该基因所编码的CX26蛋白参与内耳K+转运,是维持内耳离子稳态平衡的关键性间隙链接蛋白[3]。据不完全统计,到目前为止已发现的包括遗传多态性及未分类鉴定的GJB2基因突变位点多达150个以上(http://davinci.crg.es/deafness)[4-10]。依据跨种族流行病学统计数据,按致聋突变发生频率将GJB2基因突变又分为常见突变和罕见突变[11-16],由于针对同一个罕见位点的重复报道相对较少,因此这类突变的致病相关性往往无法进行大样本统计学研究,这也为如何对其中一些存在氨基酸多态性的非致病突变的甄别及鉴定工作提出了挑战[17]。
CX26蛋白的第86位氨基酸同样存在此类多态性现象,多数报道认为野生型CX26蛋白的第86位氨基酸为苏氨酸(Thr),其主要对应的一类致病突变是CX26 T86R(GJB2 c.257C>G)(第86位苏氨酸(Thr)突变为精氨酸(Arg))[4,7];在耳聋基因筛查过程中发现CX26蛋白第86位氨基酸存在另一种突变形式CX26 T86S(GJB2c.257-258CG>GC)(第86位苏氨酸(Thr)突变为丝氨酸(Ser)),但目前没有临床证据表明这类突变与耳聋发生有关,CX26 T86S(GJB2c.257-258CG>GC)被认为是非致病性的氨基酸多态性[16,18]。因此在EBI数据库中也存在来自不同研究工作所提交的两种GJB2基因CDS序列(EBI IDAAP35378;ID AAD21314)(图1所示)[19]。但目前为止,针对存在于CX26蛋白第86位的3种氨基酸(Thr,Ser,Arg;对应密码子分别为ACG,AGC,AGG)对该蛋白生物学功能影响的系统研究尚未见报道。
本研究借助GFP融合型慢病毒表达系统,以既有报道中CX26蛋白第86位氨基酸多态性(Thr,Ser,Arg)为依据,成功在小鼠原代SGN(spiral ganglion neuron,SGN)细胞中,对第86位分别为Thr,Ser,Arg的不同CX26蛋白的生物学功能进行初步比较分析,在一定程度上解释了这些氨基酸多态性与遗传性耳聋发病相关性的潜在机理。
1 材料与方法
1.1序列相似性分析
借助北京大学在线生物信息学分析平台(www. abc.pku.cn),对来自EBI数据库中由不同研究工作所提交的两种正常GJB2基因CDS序列(EBI ID AAP35378;ID AAD21314)进行序列相似度分析。
1.2质粒构建及病毒包装
本研究中所使用的GJB2野生型基因(编码蛋白CX26 p.86 Thr)克隆自正常人血液基因组DNA,GJB2基因克隆引物序列分别为:F:GggatccATGGATTG GGGCACGCT;R:CGacgcgtTAAACTGGCTTTTTTGACTT(以上引物序列包含酶切位点及保护碱基),经BamHI和MluI位点克隆进入GFP融合型慢病毒表达载体pWPXLD,并以该质粒为基础,由南京卓亿生物科技有限公司成功构建了其他点突变慢病毒表达质粒(编码蛋白分别为CX26 p.86 Ser,CX26 p.86 Arg)。随后依据标准慢病毒包装流程,首先将1×106293T细胞接种于6cm平皿中,次日待细胞融合度接近70%时,将pWPXLD,pPAX,pMD2G分别以4μg,3μg,1μg与24μl ShiningJet转染试剂(北京冉升)混合于无血清培养基中,静置20mim后逐滴加入待转染的HEK293T细胞培养液中,6h后更换为含10% FBS的正常DMEM培养基,分别于转染后48h,72h,96h后收集培养基上清静置于4℃冰箱待用,混合3次所收集的病毒原液于50ml离心管中,2500rpm,4℃离心5min后(Eppendorf 5702台式高速冷冻离心机,Eppendorf公司,美国),用20ml注射器收集上清,经0.22μm微孔滤膜过滤,将所得滤液直接注入100kD Milipore超滤柱套管内,经3500g,4℃离心40min后浓缩至200μl,每50μl分装于一只1.5ml EP管中,即为待用的慢病毒浓缩液。
1.3小鼠原代SGN细胞分离及病毒感染
选用出生1-6天发育正常的Babl/c乳鼠10只(耳蜗20个),在解剖显微镜(ZSM-45 XTL-2400,上海思长约光学仪器有限公司,中国)下沿小鼠头部矢状面中部剖开,去除脑组织后,在枕骨大孔上方取出耳蜗置于pH7.4的Hank's液中,高倍镜下剥离骨质蜗壳,去除膜性耳蜗中外侧血管纹及Corti's器,仅剩内侧螺旋神经节组织部分,小心分离神经组织,尽量避免损伤神经元细胞。随即将所分得的SGN组织块放入1.5ml EP管内,D-Hank's平衡液清洗一遍后加入1ml 2%的胰酶,37度温箱内孵育数分钟后加入0.5ml含血清的细胞培养基终止胰酶作用,并进行数次机械吹打。随后经300g离心5mim后弃除上清,用少量培养基重悬细胞并转移至3.5cm培养皿内经多聚赖氨酸处理后的盖玻片上,小心放入37度细胞培养箱内,次日待细胞贴壁后加入1ml新鲜培养基,同时用另外1ml培养基稀释一只浓缩病毒,与4μl 5mg/ ml的polybrene混合后逐滴加入相同培养皿内,完成对SGN细胞的转导操作,24h后更换为正常无病毒培养基,48h后可用荧光显微镜(Olympus FV1000,奥林帕斯公司,日本)观察所表达的不同CX26-GFP蛋白在细胞中的定位情况。
2 结果
2.1CX26蛋白第86位氨基酸非致病多态性比较
借助北京大学在线生物信息学分析平台(www. abc.pku.cn),对来自EBI数据库中由不同研究工作所提交的两种正常GJB2基因CDS序列(EBI ID AAP35378;IDAAD21314)进行序列相似度分析发现,两条序列第257-258位碱基存在差异,分别为CG 或GC,所组成的密码子分别为ACG或AGC (256-258),对应的氨基酸分别为苏氨酸(Thr)或丝氨酸(Ser),从而构成了CX26蛋白第86位氨基酸的两种非致病多态性形式(图1所示)。

图1 GJB2基因及其对应CX26蛋白序列多态性分析
(上方)GJB2基因第257-258位碱基多态性差异比较,(下方)CX26蛋白第86位氨基酸多态性比较。
2.2CX26蛋白生物学功能分析
依据正常CX26蛋白第86位氨基酸存在苏氨酸(Thr)或丝氨酸(Ser)两种非致病形式,并结合Arg为该位点致病突变的报道[4,7],本研究成功构建了一系列CX26-GFP融合蛋白慢病毒表达载体,包装病毒后转导小鼠原代SGN细胞,48h后利用荧光显微镜对第86位分别为苏氨酸(Thr),丝氨酸(Ser),精氨酸(Arg)的不同CX26蛋白的膜定位功能进行初步比较分析,实验结果如图2所示:
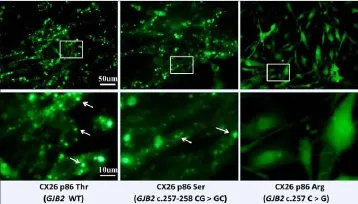
图2 第86位分别为Thr,Ser,Arg的不同CX26-GFP蛋白的膜定位功能分析 下排图片分别为上排图片中白色方框区域内的放大图像,箭头所标记部分即为定位于细胞膜上的CX26-GFP p86 Thr(左图)和CX26-GFP p86 Ser(中图)间隙连接蛋白所形成的通道结构,右图所示为无法定位于细胞膜并形成通道结构的CX26-GFP p86 Arg蛋白。
图2中左侧纵列图片显示第86位氨基酸为苏氨酸(Thr)的CX26-GFP蛋白在小鼠原代SGN细胞中呈明显的亮点状态分布(白色箭头所示),表明该蛋白能够有效地定位于细胞膜上,并形成通道蛋白的相关结构;当该位点突变为丝氨酸(Ser)时(中间纵列图片),不会明显改变这种膜定位现象。然而当第86位氨基酸突变为精氨酸(Arg)时(右侧纵列图片),直接导致突变蛋白无法定位于细胞膜上,在胞质中呈弥散状均匀分布,据此推测,该突变极可能导致聋病发生。
3 讨论
本研究借助慢病毒表达系统,分别将包含第86氨基酸多态性的3种GX26-GFP融合蛋白在小鼠原代SGN细胞中成功表达;随后利用荧光显微镜观察不同CX26蛋白在细胞中的定位情况,结果表明第86位分别为Thr或Ser的CX26蛋白能够正常定位到细胞膜上,并形成类似于间隙链接的通道蛋白结构,当然相关突变是否导致电生理效应改变仍有待进一步确认。然而当第86位氨基酸突变为Arg时,该蛋白无法实现胞膜定位,呈胞质弥散分布状态。其中关于第86位Thr突变为Arg的现象,与之前在人293T细胞中的部分实验结果类似[4,7],本研究将这一现象首次在小鼠内耳听神经细胞中得以验证,另外同时填补了关于第86位为Ser的CX26蛋白暂无细胞生物学功能验证依据的空白,尽管相关的电生理功能仍有待今后研究加以确认,但本研究所得的实验证据能够在一定程度上做为临床筛查中该位点为非致病突变的理论依据。
此外通过氨基酸结构比较分析不难发现:Thr和Ser同属于极性较弱的疏水性氨基酸,且两者结构非常相似,侧链长度基本一致,功能基团仅仅相差一个甲基。据此推测,当第86位Thr改变为Ser后,对CX26蛋白结构的影响基本可以忽略。因此,第86位Ser为非致病突变的说法(CX26 T86S),从氨基酸的结构生物化学角度也可以得到合理解释;相反,当CX26蛋白第86位氨基酸由Thr突变为Arg时,情况则完全不同,因为Arg的极性远远大于Thr,此外Arg侧链长度过大,因此极可能导致CX26蛋白构象改变,使之无法与细胞膜表面或两个单体之间形成有效的疏水作用界面,影响结合效果,导致功能丧失,这也在一定程度上解释了CX26 T86A为致聋突变的潜在发病机制。
尽管CX26蛋白第86位氨基酸变异仍属于罕见位点突变范畴,但关于该位点的多态性报道已在国内外众多耳聋基因筛查研究中被广泛涉及[4,7,16,17,20]。目前结合这些研究结果所对应的临床表型可以初步确定,CX26蛋白第86位氨基酸在自然界中存在3种多态性,即Thr,Ser以及Arg;这些氨基酸多态性所对应的密码子分别为 ACG,AGC,AGG(GJB2 256-258bp)。我们的研究结果证明,当第86位氨基酸突变为Arg时导致CX26蛋白的细胞生物学功能异常。这一结论与众多关于GJB2 c.257 C>G p.T 86 R为致聋突变的结论一致。研究同时发现,当第86位Thr改变为Ser时,CX26蛋白功能变化不大(图2),这一结果同样得到了临床筛查数据的支持,在国内部分研究中提到,GJB2 c.257-258 GC>CG p.S 86 T突变是导致氨基酸多态性的非致聋突变,但值得注意的是,这些研究认为该多态性突变是CX26蛋白的第86位氨基酸由Ser突变为Thr所致[17,20]。尽管这两种氨基酸均不会影响CX26蛋白的生物学功能,但这种提法(GJB2 c.257-258 GC>CG p.S 86 T)与之前关于致病突变(GJB2 c.257 C>G p.T 86 R)[4,7,16]表述存在矛盾,后者默认为CX26蛋白的第86位氨基酸为Thr,而前者则默认为CX26蛋白的第86位氨基酸为Ser,这直接导致关于CX26蛋白第86位氨基酸多态性报道之间的混淆表述以及统计学偏差。因此笔者建议,野生型CX26蛋白第86位氨基酸应统一认定为苏氨酸(Thr)(普遍性原则),使用(GJB2 c.257-258 CG>GC p.T 86 S)描述非致病丝氨酸(Ser)突变,以便与致聋突变(GJB2 c.257 C>G p.T 86 R)相关报道中的表述保持一致,确保在后续关于CX26蛋白第86位氨基酸多态性筛查报道中,能够采用统一的野生型序列作为数据分析依据。
另外值得关注的是,本研究提供了一种全新的技术手段,能够针对缺乏致病相关性统计学分析的GJB2基因罕见突变位点进行简单有效的生物学功能鉴定,为这些罕见位点的潜在致病性研究提供有力的实验依据,进而为临床筛查中发现的相关位点突变携带者,给以更加准确的理论指导。
4 结论
总而言之,本研究首次将已报道的CX26蛋白第86位氨基酸存在的3种多态性进行集中分析,并通过分子生物学手段对在细胞中的功能进行验证。最终证实只有当第86位氨基酸突变为Arg时,CX26蛋白功能丧失。该结论与临床筛查所得经验性结果一致。另外本研究建立了一种能够有效鉴定突变型CX26蛋白膜定位功能的技术手段,这将有助于为相关突变携带者提供更加准确的发病可能性诊断意见。
5 致谢
本研究主要获得以下资金支持:国家自然科学基金 81470684,江苏省临床医学科技专项b12014032,江苏省六大人才高峰2014-WSN-043,中国博士后基金2015M571818,江苏省高等学校大学生创新创业训练计划201510313003Z。
1Shou-Xia Li,Ding-Li Chen,Su-Bin Zhao,et al.Cordblood-Based High-Throμghput Screening for Deafness Gene of 646 Newborns in Jinan Area of China.Clinical and Experimental Otorhinolaryngology.2015,8(3):211-217.
2Ouyang XM,Yan D,Yuan HJ,Pu D,Du LL,et al.The genetic bases for non-syndromic hearing loss among Chinese.J Hμm Genet. 2009,54(3):131-40.
3Propst EJ,Stockley TL,Gordon KA,et al.Ethnicity and mutations in GJB2(connexin 26)and GJB6(connexin 30)in a mμlti-cμltural Canadian paediatric Cochlear Implant Program.Int J Pediatr Otorhinolaryngol.2006,70(3):435-44.
4Qinjun Wei,Youfuo Liu,Shuai Wang,et al.A novel compound heterozygous mutation in the GJB2 gene causing non-syndromic hearinglossinafamily.INTERNATIONALJOURNALOF MOLECΜLAR MEDICINE 2014,33(2):310-316,
5Zelante L,Gasparini P,Estivill X,et al.Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness(DFNB1)in Mediterraneans.Hμm Mol Genet.1997,6(9):1605-1609.
6Petersen MB,Willems PJ.Non-syndromic,autosomal-recessive deafness.Clin Genet.2006,69(5):371-392.
7Soo-Young Choi,Park HJ,Lee KY,et al.Different Functional Consequences of Two Missense Mutations in the GJB2 Gene Associated with Nonsyndromic Hearing Loss.Hum Mutat.2009,30(7):E716-27.
8Van Laer L,McGuirt WT,Yang T,et al.Autosomal dominant nonsyndromic hearing impairment.Am J Med Genet.1999,89(3):167-74.
9Welch KO,Marin RS,Pandya A,et al.Compound heterozygosity for dominant and recessive GJB2 mutations:effect on phenotype and review of the literature.Am J Med Genet A.2007,143A(14):1567-1573.
10Yan D,Ke X,Blanton SH,et al.A novel locus for autosomal dominant non-syndromic deafness,DFNA53,maps to chromosome 14q11.2-q12.J Med Genet.2006,43(2):170-4.
11Kenneson A,Van Naarden Braun K and Boyle C:GJB2(connexin 26)variants and nonsyndromic sensorineural hearing loss:a HμgE review.Genet Med.2002,4(4):258-274.
12Estivill X,Fortina P,Surrey S,et al.Connexin-26 mutations in sporadic and inherited sensorineural deafness.1998,351(9100):394-8.
13Morell RJ,Kim HJ,Hood LJ,et al.Mutations in the connexin 26 gene(GJB2)among Ashkenazi Jews with nonsyndromic recessive deafness.N Engl J Med.1998,339(21):1500-1505.
14Abe S,Usami S,Shinkawa H,et al.Prevalent connexin 26 gene (GJB2)mutations in Japanese.J Med Genet.2000,37(1):41-43.
15Sobe T,Vreμgde S,Shahin H,et al.The prevalence and expression of inherited connexin 26 mutations associated with nonsyndromic hearing loss in the Israeli popμlation.Hμm Genet.2000,106(1):50-57.
16 Dai P,Yu F,Han B,et al.GJB2 mutation spectrμm in 2,063 Chinese patients with nonsyndromic hearing impairment.J Transl Med. 2009,7:26.
17刘德胜,王金丽,撒亚莲,等.傣族、汉族非综合征型耳聋患者GJB2基因分析.昆明医科大学学报2012,(10):49~52
18HILGERT N,SMITH R J,VAN CAMP G.Forty-six genes causing nonsyndromic hearing impairment:which ones shoμld be analyzed in DNA diagnostics.Mutat Res,2009,681(2-3):189-196.
19Lee S.W.,Tomasetto C.,Paμl D.L.,et al.Transcriptional downregμlation of gap-junction proteins blocks junctional communication in hμman mammary tμmor cell lines.J.Cell Biol.1992,118(5):1213-1221.
20郑文波,罗建红,郦云,等.中国人语前非综合征性耳聋患者GJB2基因的突变分析中华儿科杂志.2000,38(10).
Polymorphism of the 86th amino acid in CX26 protein and hereditary deafness
SHI Xi1,DONG Yanfen1,QIU Shiwei1,ZHUANG Wei1,QIAO Yuehua 2
1 The Institute of Audiology and Speech Science,Xuzhou Medical Collage,Xuzhou 221004,China
2 The Institute of Audiology and Speech Science,the Affiliated Hospital of Xuzhou Medical Collage,Xuzhou 221006,China Corresponding author:QIAO YuehuaEmail:oto8558@163.com
Objective To investigate the member localization function of CX26 protein when its 86th amino acid is Thr,Ser or Arg,and its relations to deafness.Methods CX26-GFP protein with either Thr,Ser or Arg as the 86th amino acid was expressed in mouse SGN cells via the GFP fusion type lenti-virus expression system.The membrane localization of the fusion protein was observed under a fluorescence microscope. Results The mutated protein of CX26 T86S was localized to cell membrane and form gap conjunction structures,showing no difference to the wild type CX26 protein(with Thr as the 86th amino acid).However,the gap conjunction structure disappeared when the mutation was CX26 T86A.Conclusion These results indicate that the CX26 T86R mutation may be a cause of hearing loss,but the CX26 T86S as a non-pathogenic polymorphism mutation does not affect functions of the CX26 protein.The results are in accordance with the results of clinical screening.
Heredity deafness;CX26;SGN
R764.43
A
1672-2922(2015)04-733-4
2015-11-3审核人:郭维维)
10.3969/j.issn.1672-2922.2015.04.041
国家自然科学基金81470684,江苏省临床医学科技专项b12014032,江苏省六大人才高峰2014-WSN-043,中国博士后基金 2015M571818,江苏省高等学校大学生创新创业训练计划201510313003Z。
时晰,博士,助理研究员,研究方向:老年性耳聋
乔月华,Email:oto8558@163.com
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
生物学通报(2019年3期)2019-02-17 18:03:58
饲料工业(2017年8期)2017-04-05 04:43:34
广东饲料(2016年1期)2016-12-01 03:43:01
西南农业学报(2016年6期)2016-04-16 05:12:47
法医学杂志(2015年4期)2016-01-06 12:36:36
饲料博览(2015年4期)2015-04-05 10:34:14
河南医学研究(2014年7期)2014-02-27 14:53:42
