
酮康唑对大鼠鼻腔和灌胃给予咪达唑仑及其代谢产物药动学差异的影响
2015-08-31 09:59:04王晓英李敬来郑爱萍张振清庄笑梅抗毒药物与毒理学国家重点实验室军事医学科学院毒物药物研究所北京100850
中国药理学与毒理学杂志 2015年6期
王 娟,王晓英,李敬来,李 峥,郑爱萍,张振清,庄笑梅(抗毒药物与毒理学国家重点实验室,军事医学科学院毒物药物研究所,北京 100850)
酮康唑对大鼠鼻腔和灌胃给予咪达唑仑及其代谢产物药动学差异的影响
王 娟,王晓英,李敬来,李 峥,郑爱萍,张振清,庄笑梅
(抗毒药物与毒理学国家重点实验室,军事医学科学院毒物药物研究所,北京 100850)
目的在系统比较大鼠鼻腔和灌胃给予咪达唑仑药动学特征的基础上,进一步比较酮康唑对2种给药途径药动学的影响。方法 24只大鼠随机分为4组,每组6只。其中2组分别经鼻腔或灌胃只给予咪达唑仑(1 mg·kg-1),另外2组联用细胞色素P450酶3A(CYP3A)抑制剂酮康唑(30 mg·kg-1)后再分别经鼻腔或灌胃给予咪达唑仑,不同时间点采集血样,测定咪达唑仑和1′-羟基咪达唑仑浓度,计算药代动力学参数,并进行统计学分析。结果 大鼠单独经鼻腔和灌胃给予咪达唑仑后,原型药物的达峰时间(Tmax)分别约为2和25 min,药时曲线下面积(AUC)分别为296和179 μg·L-1·h。合用酮康唑后,在鼻腔和灌胃给药条件下,咪达唑仑原型药物在大鼠体内的AUC分别增加到原来的2.1和3.3倍。但是,酮康唑不改变咪达唑仑鼻腔给药的Tmax,而合用酮康唑后,咪达唑仑灌胃给药的Tmax延长至1.14h。结论 咪达唑仑经鼻腔给药与经口服给药相比,吸收迅速、药物暴露量大,更适合于临床急救。咪达唑仑经鼻腔给药合用酮康唑后,不改变吸收速度,但抑制其代谢转化,药物体内驻留时间明显延长;咪达唑仑口服给药合并给予酮康唑后,吸收减慢,抑制代谢转化,体内药物暴露量明显增加。由于咪达唑仑的中枢镇静作用,2种途径合用酮康唑时均应考虑适当减少给药剂量或延长给药间隔。
咪达唑仑;酮康唑;药代动力学;药物相互作用
DOl:10.3867/j.issn.1000-3002.2015.06.010
癫痫发作是临床急危症之一,需要立即抢救,若不及时控制可造成脑组织神经系统的不可逆损害,严重时可危及生命。尽早控制惊厥持续发作是挽救生命改善预后的关键[1]。咪达唑仑(midazolam,MDZ)为环状结构的苯二氮卓艹类化合物,是一种新型苯二氮卓艹类药物,具有抗焦虑、肌松、镇静和催眠等作用,作为麻醉剂广泛应用于临床[2]。后来在动物实验中发现,其具有显著的抗惊厥作用,特别是当惊厥不能为地西泮、苯妥英钠和苯巴比妥所控制时MDZ仍然有效。MDZ不但能成功地控制惊厥发作,而且能够有效地消除发作间期痫样放电[3]。MDZ是细胞色素P450酶3A(cytochrome P-450 3A,CYP3A)的经典底物,人体内主要产物为1′-羟基咪达唑仑(1′-OH-MDZ);大鼠体内产物除了1′-OH-MDZ外,还有4-OH-MDZ。1′-OH-MDZ的活性约为MDZ的1/10[4]。因此,原型药物暴露量的变化是药效改变的决定性因素。
虽然MDZ对惊厥和癫痫的稳定疗效已受到临床医师的广泛认可,鼻腔给药途径也是临床常用的途径[5],但MDZ经鼻腔给药和口服给药的药代动力学差异未见报道。CYP3A介导的药物相互作用是否会因给药途径不同而不同也值得探索。本研究的目的包括:①系统比较大鼠鼻腔或ig给予MDZ后,原型药物及1′-OH-MDZ的药动学特征及差异;②合用CYP3A抑制剂酮康唑(ketoconazole,KTZ)后,观察KTZ对MDZ经以上2种途径给药后,体内药动学差异的影响。
1 材料与方法
1.1 药品和试剂
MDZ鼻喷剂(25 g·L-1,批号20121119-1),由军事医学科学院毒物药物研究所药物制剂研究室提供;MDZ注射液(5 g·L-1,批号20110802),购自江苏恩华药业股份有限公司。MDZ标准品(纯度>99%,批号MD101102),宜昌人福药业有限责任公司;1′-OH-MDZ标准品(纯度>99%),美国BD Gentest公司;KTZ标准品(纯度>98%,批号SLBG3774V)美国Sigma公司;内标盐酸苯环壬酯(8021)(批号010626,纯度99.8%),白色粉末,由军事医学科学院毒物药物研究所药物化学合成室提供;乙晴和甲醇,色谱纯,购于Fisher Scientific(中国)公司;实验用水为娃哈哈纯净水,娃哈哈集团有限公司;甲酸,分析纯,北京化学试剂公司;氯化钠注射液(0.9%),山东华鲁制药有限公司。
1.2 仪器
美国Finnigan公司TSQ Quantum Discovery MAX型液质联用系统(liquid chromatography tandem mass spectrometry,LC-MS/MS)。控制软件为Xcalibur1.4,质谱数据分析采用Lcquan2.0数据处理系统。色谱柱,BetaBasic-18(50 mm× 2.1 mm,5 μm,Thermo Electron Corporation,美国)。
1.3 动物及分组处理
成年SD大鼠24只,雌雄各半,体质量为180~220 g,由军事医学科学院动物中心提供,动物合格证号:SYXK-(军)2011-0010。
24只大鼠随机分为4组,每组6只,雌雄各半。Ⅰ组:ig给予KTZ(30 mg·kg-1)30 min后,再给予MDZ鼻喷制剂(1 mg·kg-1);Ⅱ组:ig等量的溶媒30 min后,给予MDZ鼻喷制剂(1 mg·kg-1);Ⅲ组:ig给予KTZ(30 mg·kg-1)30 min后,再ig给予MDZ溶液剂(1 mg·kg-1);Ⅳ组:ig等量的溶媒30 min后,再ig给予MDZ溶液剂(1 mg·kg-1)。分别于给药前和给药后2,5,10,15,20,30,45 min,1,2,3,4,6,8 和24 h,由眼眶静脉取血约0.2 mL,置于含有肝素的抗凝试管中,离心(3500×g,4℃)15 min分离血浆,准确取血浆0.05 mL 2份,置-80℃冰箱冻存备测。随行标准曲线浓度范围:MDZ 2.0~2000.0 μg·L-1,1′-OHMDZ 0.2~200.0 μg·L-1。含MDZ/1′-OH-MDZ浓度为4.0/0.4,100.0/10.0,1500.0/150.0 μg·L-1的大鼠空白血浆样品作为质控样品。
1.4 色谱条件
流动相组成:水∶乙腈(含0.1%甲酸)=85∶15 (V∶V)。流速为0.45 mL·min-1,进样量10 μL,运行时间为4.5 min,柱温25℃。
1.5 质谱条件
选择大气压化学电喷雾源(ESI),鞘气45 psi,辅助气12 psi,喷雾电压4800 V,温度320℃,源内碰撞诱导裂解(in-source CID)-10 V。MDZ,1′-OH-MDZ和内标检测的质荷 比(m/z)分别为326.1,342.1和358.0。以选择性反应离子(SRM)方式在正离子模式下检测m/z 326.1的碎片离子为m/z 222.0;m/z 342.1的碎片离子为m/z 168.0;m/z 358.0的碎片离子为m/z 156.0。
1.6 血浆样品处理和测定
血浆样品0.05 mL加入0.1 mL内标(500 μg·L-1,溶剂为乙腈)和乙腈0.05 mL,涡旋1 min,离心(12 000×g,4℃)10 min,取上清0.15 mL于进样瓶内,进样分析。
1.7 数据处理及统计学分析
采用Winnolin药代动力学程序对所测数据进行c-t曲线拟合,以非房室模型(统计矩法)计算主要药代动力学参数:药物峰浓度(cmax)、药时曲线下面积(AUC)、达峰时间(Tmax)、半衰期(t1/2)和平均驻留时间(MRT)。采用SAS统计软件对各组参数进行双侧成组t检验。
2 结果
2.1 同时测定大鼠血浆中MDZ和1′-OH-MDZ的LC-MS/MS方法验证
MDZ、1′-OH-MDZ和内标的色谱峰(典型色谱图略)均尖锐对称,且分离度良好,血浆中内源性物质不干扰样品的分离测定,该方法具有较高的专属性。MDZ的大鼠血浆定量范围在2.0~2000.0 μg·L-1内时,呈良好的线性关系,回归方程为Y(Ai/As)=0.008+ 0.009X(Ci),相关系数的平方γ2为0.9928,定量下限为2.0 μg·L-1,1′-OH-MDZ的血浆定量范围在0.2~200.0 μg·L-1内时,呈良好的线性关系,回归方程为Y(Ai/As)=-0.0007+0.0057X(Ci),相关系数的平方γ2为0.9961,定量下限为0.2 μg·L-1。MDZ/1′-OH-MDZ浓度为4.0/0.4,100.0/10.0,1500.0/150.0 μg·L-1的质控样品在大鼠血浆中的回收率均在93.9%以上;批内和批间的精密度分别在13.2%和13.0%以内;其准确度控制在89.3%和106.1%以内;MDZ和1′-OH-MDZ的基质效应影响范围分别为72.4%~80.3%和111%~128%,大鼠血浆对MDZ和1′-OHMDZ的基质效应出现了较明显的抑制和增强,但由于变异较小(RSD在3.9%~9.8%之间),不影响样品定量准确性[6]。以上验证结果说明,该方法满足生物样品定量检测要求。
2.2 大鼠经鼻腔和灌胃给予MDZ后药代动力学特征比较
大鼠经鼻腔和灌胃给予MDZ后,原型药物及代谢产物1′-OH-MDZ的药时曲线见图1,相应的药代动力学参数见表1。
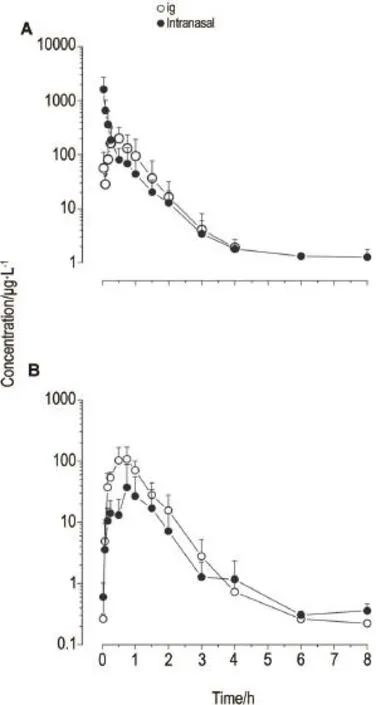
Fig.1 Mean plasma concentration-time profiles of midazolam(MDZ)(A)and 1′-hydroxymidazolam(1′-OHMDZ)(B)after intranasal administration and ig of MDZ (1 mg·kg-1)in rats.±s,n=6.
表1结果显示,MDZ(1 mg·kg-1)经鼻腔给予大鼠后,原型药物被吸收入血的速度明显快于ig给药,cmax也明显高于ig给药,虽然AUC未显示统计学差异,但也是ig给药的1.6倍左右。此外,与ig给药相比,MDZ经鼻腔给药在大鼠体内的代谢率降低,AUC(1-OH-MDZ)/AUC(MDZ)比值在鼻腔给药时为16%,而ig给药时为66%。
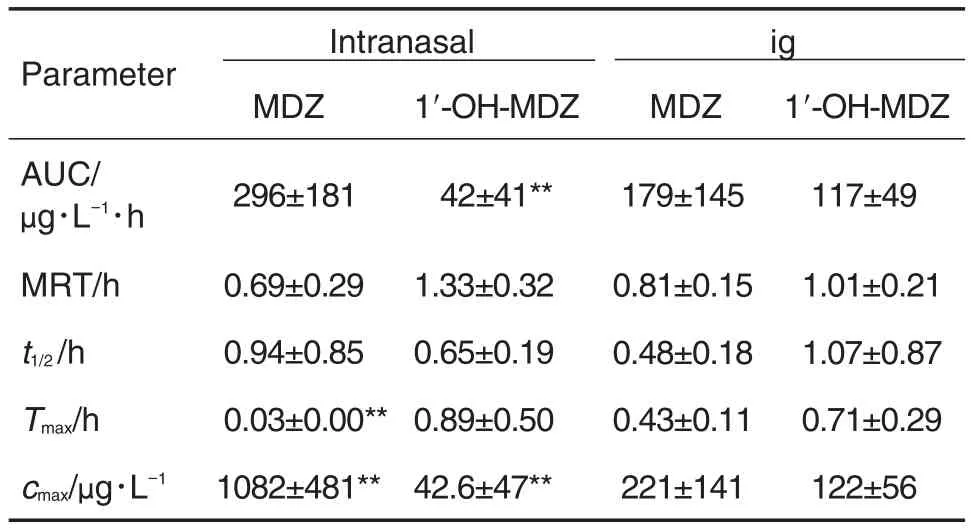
Tab.1 Pharmacokinetic parameters of MDZ and its metabolite 1′-OH-MDZ determined by LC-MS/MS following intranasal or ig of MDZ(1 mg·kg-1)in rats
2.3 KTZ预处理组大鼠经鼻腔和灌胃给予MDZ后药代动力学特征比较
大鼠先ig给予KTZ 30 min后,再分别ig(药时曲线见图2)及经鼻腔(药时曲线见图3)给予MDZ (1 mg·kg-1)(MDZ及1′-OH-MDZ;相应的药代动力学参数见表2,计算KTZ对MDZ药动学参数改变倍数(表3)。
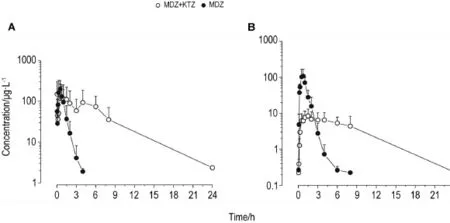
Fig.2 Mean concentration-time profile of MDZ(A)and 1′-OH-MDZ(B)following ig of MDZ(1 mg·kg-1)with or without KTZ(30 mg·kg-1)in rats.±s,n=6.
表2结果显示,大鼠先经ig给予KTZ (30 mg·kg-1)后,再分别经鼻腔和ig给予MDZ (1 mg·kg-1),原型药物在体内的暴露量几乎相当,但经鼻腔吸收入血的速度未受影响,仍明显快于ig给药。表3结果显示,合用KTZ后,2种给药途径均增加了MDZ原型药物的体内暴露量,而代谢产物量均未增加,原型药物的清除速率都明显降低(MRT增加了4倍)。表4结果进一步证明,大鼠单独给予MDZ时,ig给药后产物与原药的比例(66%)明显高于鼻腔给药(16%)。合用酮康唑后,对ig给予MDZ的代谢酶抑制作用非常明显(产物与原药的比例从66%降低到19%),统计学检验有显著性差异。但鼻腔给药后,虽然产物与原药的比例从16%降低到7%,但是统计学分析无显著性差异。
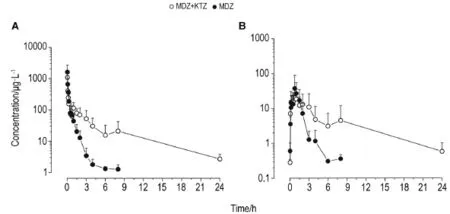
Fig.3 Mean concentration-time profile of MDZ(A)and 1′-OH-MDZ(B)following intranasal of MDZ(1 mg·kg-1)with or without KTZ(30 mg·kg-1)in rats.±s,n=6.
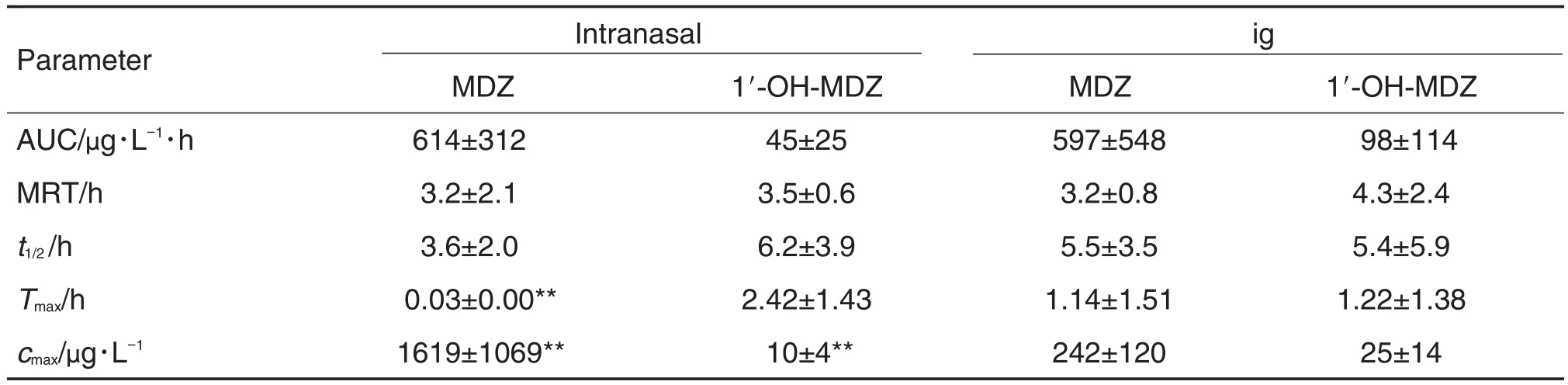
Tab.2 Pharmacokinetic parameters of MDZ and 1′-OH-MDZ determined by LC-MS/MS following intranasal or ig of MDZ(1 mg·kg-1)after coadministration with ketoconazole(KTZ)(30 mg·kg-1)in rats
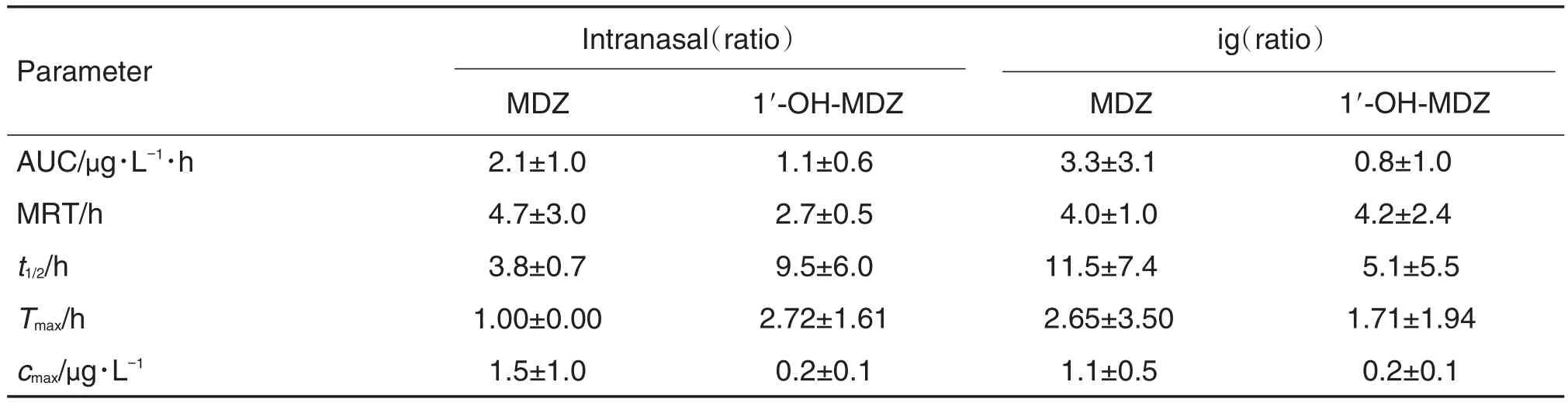
Tab.3 Ratios of pharmacokinetic parameters of MDZ and 1′-OH-MDZ following intranasal or ig of MDZ(1 mg·kg-1)after coadministration with(30 mg·kg-1)to the parameters of the administration without KTZ in rats
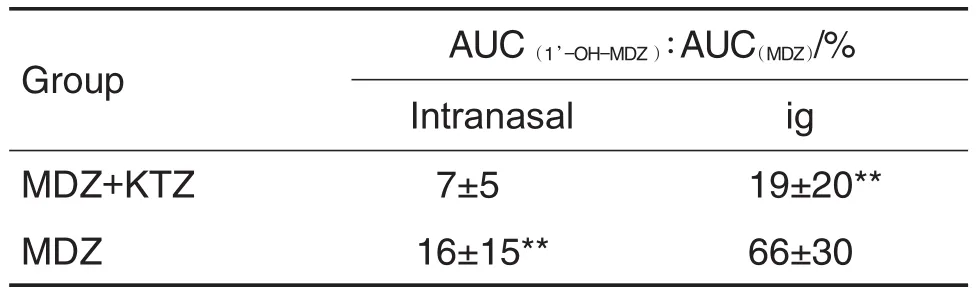
Tab.4 Percentage of AUC(1’-OH-MDZ)/AUC(MDZ)following ig or intranasal of MDZ(1 mg·kg-1)after coadministration with or without KTZ in rats
3 讨论
为了研究MDZ经不同途径给药后,在大鼠体内的药代动力学过程,特别是观察CYP3A代谢酶抑制剂对MDZ体内的影响,需要建立同时检测大鼠血浆中MDZ及主要代谢产物1′-OH-MDZ的方法。由于MDZ是经典的CYP3A底物,其检测方法的报道很多。本研究中建立的方法与文献相比,检测范围相当(MDZ 1~1000 μg·L-1,1′-OH-MDZ 1~100 μg·L-1)[7]。经方法学验证后,开展了本研究。
大鼠分别经鼻腔和ig给予相同剂量的MDZ后,原型药物和代谢产物1′-OH-MDZ的药动学特征显示出一定的差异。经鼻腔给药后,MDZ约2 min即达峰值,ig给药的达峰时间约为25 min(与文献报道的30 min一致),由于在癫痫和惊厥发作救治时,MDZ能否迅速发挥药效是十分重要的[8],相比之下,鼻腔给药途径更适合于临床应用。
由于MDZ是CYP3A的特异底物,临床上很多药物是CYP3A的抑制剂[9],临床观察发现合用CYP3A的抑制剂可以延长MDZ的镇静时间[10]。考虑到MDZ经鼻腔和ig给药后药代动力学特征有明显的差别,合用CYP3A抑制剂后对MDZ药代动力学的影响是否有差异也值得研究。结果显示,合用KTZ,两种给药途径给予MDZ后,原型药物在体内的暴露量都出现了明显的增加,口服给药组的增加程度大于经鼻腔给药,并且在目前的给药剂量下,2组的总体暴露量相当(614与597 μg·L-1·h-1),提示当肝和肠道CYP3A的功能均被抑制后,2种给药途径的生物利用度是相当的。从MDZ和代谢产物1′-OH-MDZ的AUC变化规律可以看出,合用CYP3A抑制剂后,主要抑制了MDZ的代谢转化,产物的生成量相对于原型药物均出现了降低(鼻腔给药的从16%降低到7%,ig给药的从66%降低到19%)。除此之外,合用CYP3A的抑制剂KTZ后,MDZ经鼻腔给药组的达峰时间没有改变,仍约为2 min,而经ig给药组达峰时间推迟到1 h左右。上述结果提示,合用KTZ等CYP3A抑制剂后,MDZ经鼻腔途径给药对药效发挥的速度没有改变,但KTZ对其鼻腔给药的药效维持时间可能会明显延长4倍左右;ig途径给药会影响药效发挥速度,KTZ对其ig给药的药效维持时间可能也会明显延长3~4倍。
MDZ虽然是安全窗范围较大的药物[11],代谢产物的活性也明显低于原型药物,但是合用CYP3A抑制剂后,代谢转化的抑制也会在一定程度上影响药效发挥的时间。本研究结果提示,MDZ经鼻腔给药吸收迅速,适合于临床急救。合用CYP3A抑制剂后,鼻腔给药不改变药物吸收速度,但抑制其代谢转化,体内驻留时间明显延长,暴露量增加。ig给药合并给予KTZ后,吸收减慢,抑制代谢转化,体内药物暴露量明显增加。由于MDZ的中枢镇静作用,合并用药时均应考虑适当减少给药剂量或增加给药时间间隔,确保临床安全合理用药。
[1]Wu YZ,Cheng XM,Yu LZ,Ouyang K.Efficacy of 34 cases of midazolam treatment of children with convulsive status epilepticus[J].China Prac Med(中国实用医药),2010,5(14):172-173.
[2]Zhong JM,Li JH,Chen Y.Continuous intravenous infusion of midazolam treatment of children with status epilepsy[J].Chin J Pediatr(中华儿科杂志),2004,42(4):299-300.
[3]Zhu J,Zhang YQ,Zhang PY,Li H,Liu XJ.Clinical effectiveness of intranasal midazolam administration in acute children seizures control[J].Chin J Pract Pediatr(中国实用儿科杂志),2009,24(4):300-301.
[4]Duan YX,Li Q,Zheng AP,Zhu XW.Pharmacokinetics and the absolute bioavailability of midazolam rectal gel in rabbits[J].J Int Pharm Res(国际药学研究杂志),2013,40(3):338-343.
[5]Gilat E,Goldman M,Lahat E,Levy A,Rabinovitz I,Cohen G,et al.Nasal midazolam as a novel anticonvulsivetreatmentagainstorganophosphateinduced seizure activity in the guinea pig[J].Arch Toxicol,2003,77(3):167-172.
[6]Zhuang XM,Yuan M,Zhang ZW,Wang XY,Zhang ZQ,Ruan JX.Determination of 4-dimethylaminophenol concentrations in dog blood using LC-ESI/MS/MS combined with precolumn derivatization[J].J Chromatogr B Analyt Technol Biomed Life Sci,2008,876(1):76-82.
[7]Liu CH,Huang XT,Zheng X,Li N,Li YY,Mi SQ,et al.LC-MS/MS determination of midazolam and 1′-hydroxymidazolam,its main metabolite in rat plasma and its pharmacokinetics[J].J Guangdong Pharma Univ(广东药学院学报),2012,28(1):61-64.
[8]Liu ZG.A comparative study on the efficacyof midazolam solution administered nasally and diazepam solution administeredrectally in seizure control in children[J].Pediatr Emerg Med(小儿急救医学),2004,11(3):152-153.
[9]Zhao YH,Tian AY,Ma H,Wang JK.Contribution of CYP3A4 to catalysis of ketamine in human hepatic microsome[J].Chin Pharmacol Bull(中国药理学通报),2012,28(7):986-988.
[10]Ding LF,Hong YM,Jing LP,Dong WL.Determination of Midazolam in mouse plasma serum concentration distribution by reversed-phase HPLC [J].Chin Pharm J(中国药学杂志),2011,46(24):1943-1945.
[11]Wang SJ,Zhu P,Xue HR.The clinical study on treating infantile febrile seizures with midazolam nasal drip[J].J Pediatr Pharm(儿科药学杂志),2005,11(3):37-38.
(本文编辑:齐春会 沈海南)
Effect of ketoconazole on pharmacokinetics of midazolam and its metabolite through intranasal and intragastric routes in rats
WANG Juan,WANG Xiao-ying,LI Jing-lai,LI Zheng,ZHENG Ai-ping,ZHANG Zhen-qing,ZHUANG Xiao-mei
(State Key Laboratory of Toxicology and Medical Countermeasures,Institute of Pharmacology and Toxicology,Academy of Military Medical Sciences,Beijing 100850,China)
OBJECTlVE To investigate the effect of ketoconazole on the pharmacokinetic(PK)behaviors of midazolam and its metabolite through intranasal and intragastric(ig)routes in rats.METHODS Twenty-four rats were evenly divided into 4 groups.Two groups of rats were administrated singly with midazolam(1 mg·kg-1)through intranasal or ig route.The other two groups were concomitant with CYP3A inhibitor,ketoconazole(30 mg·kg-1),midazolam(1 mg·kg-1)through the same two routes. Blood samples were collected from different time points.Plasma concentration of midazolam and 1′-hydroxymidazolam was determined.Major pharmacokinetic parameters were calculated and statistical tests were performed by using t test.RESULTS Tmaxwas about 2 and 25 min for rats administered singly with midazolam via intranasal or ig routes,respectively and AUC was 296 and 179 μg·L-1·h,respectively.When concomitant with ketoconazole,AUC increased to 2.1 and 3.3 folds the original value for intranasal and ig routes,respectively.However,the Tmaxvalue of midazolam via intranasally didn′t change after being coadministrated with ketoconazole,but Tmaxincreased to 1.14 h via ig.CONCLUSlON Compared with administration via ig,intranasal route administrated midazolam displays significant advantages of faster absorption and higher exposure,which are vital for the first aid.Concomitant with CYP3A inhibitor and midazolam via intranasal route,the absorption speed is not affected,but with the metabolism blocked,the systemic exposure is greatly elevated.While via ig,both absorption speed and metabolism are inhibited.The dose should be cut down or the dosing interval increased in clinic practice in this concomitant situation.
midazolam;ketoconazole;pharmacokinetics;drug interactions
The project supported by National Science and Technology Major Project(2012ZX09301003-001-009);National Science and Technology Major Project(2013ZX09J13103-01B);National Science and Technology Major Project(2014ZX09507001003);and National Science and Technology Major Project(2014ZX09J14103-01A)
ZHUANG Xiao-mei,E-mail:xiaomeizhuang@163.com
R969.2
A
1000-3002(2015)06-0939-06
国家科技重大专项(2012ZX09301003-001-009);国家科技重大专项(2013ZX09J13103-01B);国家科技重大专项(2014ZX09507001003);国家科技重大专项(2014ZX09J14103-01A)
王 娟,学士,助理实验师,主要从事药物代谢研究。
庄笑梅,E-mail:xiaomeizhuang@163.com
(2015-02-09接受日期:2015-11-12)
猜你喜欢
化工管理(2022年14期)2022-12-02 11:44:48
昆明医科大学学报(2021年6期)2021-07-31 07:40:22
中国民间疗法(2021年4期)2021-06-09 09:20:08
中华养生保健(2020年3期)2020-11-16 00:53:20
广东医科大学学报(2020年4期)2020-08-24 07:11:20
中国医院院长(2017年7期)2017-06-15 12:58:15
国外医药(抗生素分册)(2016年3期)2016-07-12 14:25:09
中国卫生标准管理(2015年5期)2016-01-14 05:17:05
中国药业(2014年20期)2014-05-17 03:13:41
中成药(2014年9期)2014-02-28 22:28:49
