
吗啡急性处理小鼠的微阵列数据分析和生物标记识别
2015-08-22 08:47张文华王燕平
天津医药 2015年2期
张文华,王燕平
吗啡急性处理小鼠的微阵列数据分析和生物标记识别
张文华,王燕平△
目的通过生物信息学方法,识别吗啡急性处理小鼠的差异表达基因及其富集的通路。方法从高通量基因表达数据库(Gene Expression Omnibus)下载吗啡急性处理小鼠的微阵列数据,并调用R语言3.1.0软件的质量控制包AffyQCReport 1.42.0对数据样本进行质量控制分析,运用R语言的微阵列数据线性模型识别吗啡处理前后的差异表达基因,采用表达分析系统检测算法进行差异表达基因的通路富集分析。结果吗啡急性处理小鼠的微阵列基因表达数据具有良好的均一性和阵列强度相似性,识别得到Gm11627、Zfand4、Zbtb16、Pkp2和Plin4等481个差异表达基因和癌症、黑素原生成和丝裂原活化蛋白激酶信号等8条代谢通路。结论所识别的差异表达基因和代谢通路成为吗啡急性处理小鼠潜在的生物标记。
吗啡;小鼠;微阵列分析;基因表达;生物学标记;功能富集分析
吗啡具有镇痛、止咳、降低血压等作用[1]。同时,吗啡的过量吸入会形成神经系统分子和细胞的适应性,进而导致对药品的耐受、物理依赖和成瘾[2]。因此,深入研究吗啡对人体的生理作用机制,有助于对其高效地利用。生物信息学从核酸和蛋白质序列出发,分析序列中包含的结构功能的生物信息,可部分代替大量的分子生物学工作,具有快捷、高效、覆盖面广等优势[3]。本研究通过微阵列芯片数据的基因表达情况分析吗啡对小鼠组织进行急性处理前后基因表达的差异,并对差异表达基因进行代谢通路的富集分析,为研究吗啡对人体的作用机制提供参考。
1 材料与方法
1.1基因表达谱数据本文从高通量基因表达数据库(Gene Expression Omnibus)[4]下载E-GEOD-7762[2]数据样本表达微阵列数据,采用Mouse430_2平台进行检测,分析流程见图1。同时从数据库下载原始raw文件以及该平台探针注释信息文件。实验选用来自美国缅因州巴尔港的Jackson实验室的4种不同种属的小鼠(Mice),其种名分别为129P3/J、DBA/2J、C57BL/6J和SWR/J。该微阵列数据实验组和对照组分别选用12只小鼠为实验对象,每个种属各3只,鼠龄8~10周,体质量20~30 g,雌雄不限。吗啡注射以前,2组小鼠均等量生理盐水注射4 d,3次/d,食物与水正常喂养。实验组以20 mg/kg一次性皮下注射吗啡,4 h以后取小鼠组织样品进行分析;对照组为等量生理盐水注射,4 h后取小鼠组织样品[2]。
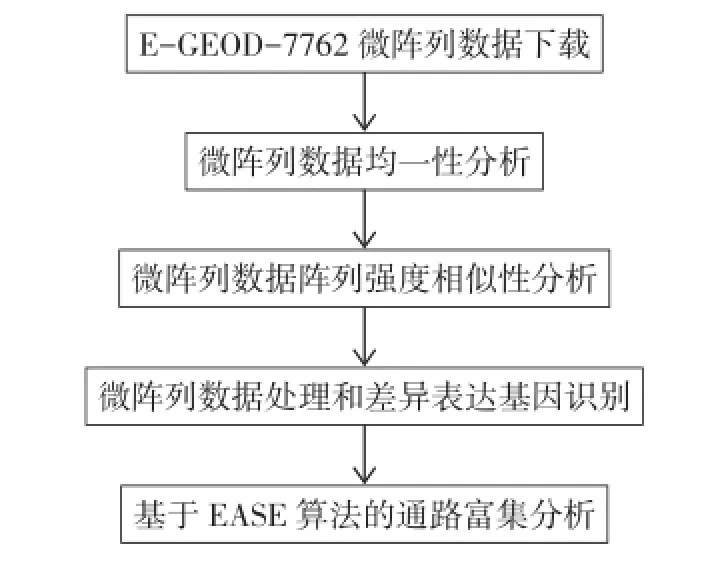
Fig.1 The experimental protocol of microarray data on mice who were acutely administered with morphine图1 吗啡急性处理小鼠微阵列数据分析流程
1.2微阵列数据均一性分析调用R语言(R programming language)3.1.0软件的质量控制包AffyQCReport 1.42.0进行微阵列数据的均一性分析。使用该质量控制平台读取阵列数据文件,对微阵列数据进行质量评估,得到样本数据的厢式图、趋势图和质量控制统计图,以此衡量微阵列数据的均一性[5]。actin3/actin5基因的尺度因子<3,且actin3/actin5内参基因的尺度因子在-1和1之间,则为芯片数据均一性良好,以线条及端点表示[6-7]。
1.3微阵列数据阵列强度相似性分析调用R语言3.1.0软件的质量控制包AffyQCReport 1.42.0进行微阵列数据的阵列强度相似性分析,相关系数大于0.90为相似性良好[8]。将微阵列数据输入质量控制平台进行质量评估,由R语言输出该样本数据的阵列强度相似性报告。
1.4差异表达基因识别首先读取全部芯片基因数据并进行预处理。输入原始数据,并对所有样本表达数据进行背景矫正。删除没有对应基因的探针,同时调用R语言3.1.0的geneFilter包,运行其中的featureFilter函数以过滤对应有多个探针的基因,得到部分基因[9]。对上述预处理所得基因进行进一步筛选,运用R语言的微阵列数据线性模型(linear models for microarray data,LIMMA)提取基因表达量差异倍数值大于2的基因[10]。并校正假阳性率(False discovery rate)<5%和P<0.05,得到相关的差异表达基因[11]。
1.5通路富集分析本文采用表达分析系统检测算法(ex⁃pression analysis systematic explorer,EASE)进行京都基因与基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)通路富集在线分析[12-13]。基于EASE算法的KEGG通路富集分析通过生物功能信息学微阵列分析注释(DAVID)网站[14]实现。在KEGG通路分析过程中,提交所得的481个差异表达基因,并设置P<0.05和通路最小基因数=2作为差异表达基因的富集条件,以筛选差异表达基因富集程度较高的通路。
2 结果
2.1微阵列数据均一性分析2组全部数据的actin3/actin5的尺度因子均<2,且内参基因actin3/actin5的尺度因子在-1~1之间,芯片数据均一性良好,见图2。
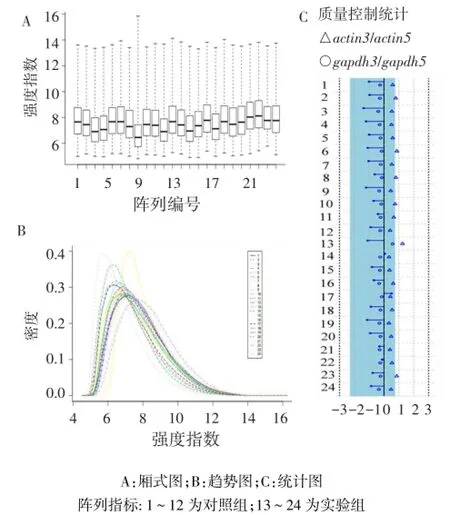
Fig.2 Intension similarity of microarray data of E-GEOD-7762图2 E-GEOD-7762微阵列芯片数据阵列强度相似性
2.2微阵列芯片数据阵列强度相似性分析2组的数据相似性均>0.90,整体阵列强度相似性良好,见图3。
2.3差异表达基因识别本文共读取45 101条芯片基因数据,其中10 313条探针基因,最终识别得到481个差异表达基因。差异表达基因的火山图,见图4。P值较小的前20个差异表达基因,见表1(仅列出NCBI官方基因简称,以P值排序)。微阵列芯片数据与前20个差异表达基因的表达调节热图,见图5。其中P值较小的Zbtb16、Plin4、Pkp2、Cebpa、Gm11627和Zfand4等差异表达基因在实验组的表达量显著增大。
2.4通路富集分析吗啡急性处理小鼠后,差异表达基因主要富集于癌症、黑素原生成和丝裂原活化蛋白激酶信号等8条通路,以P值排序,见表2。
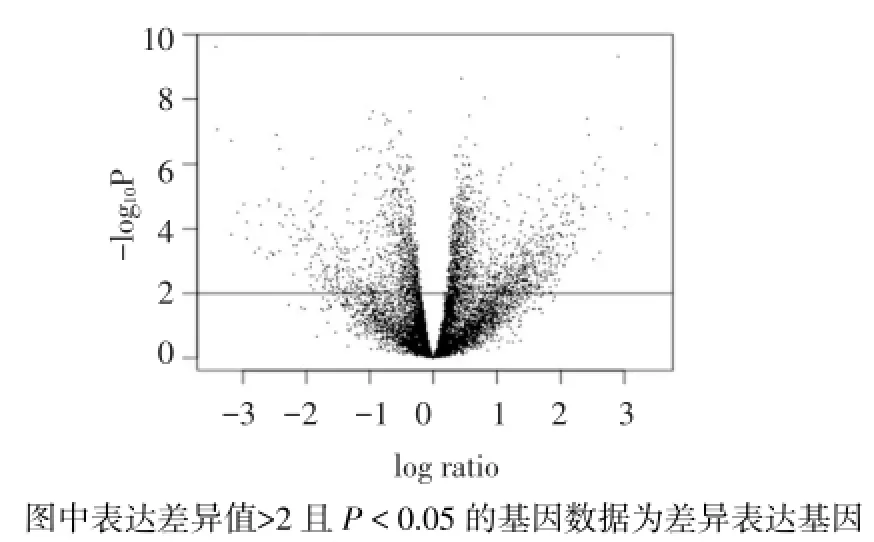
Fig.4 Volcano plot for P-value versus ratio for each gene samples including subjects and normal samples图4 2组所有基因数据的P值与表达差异值对应关系火山图
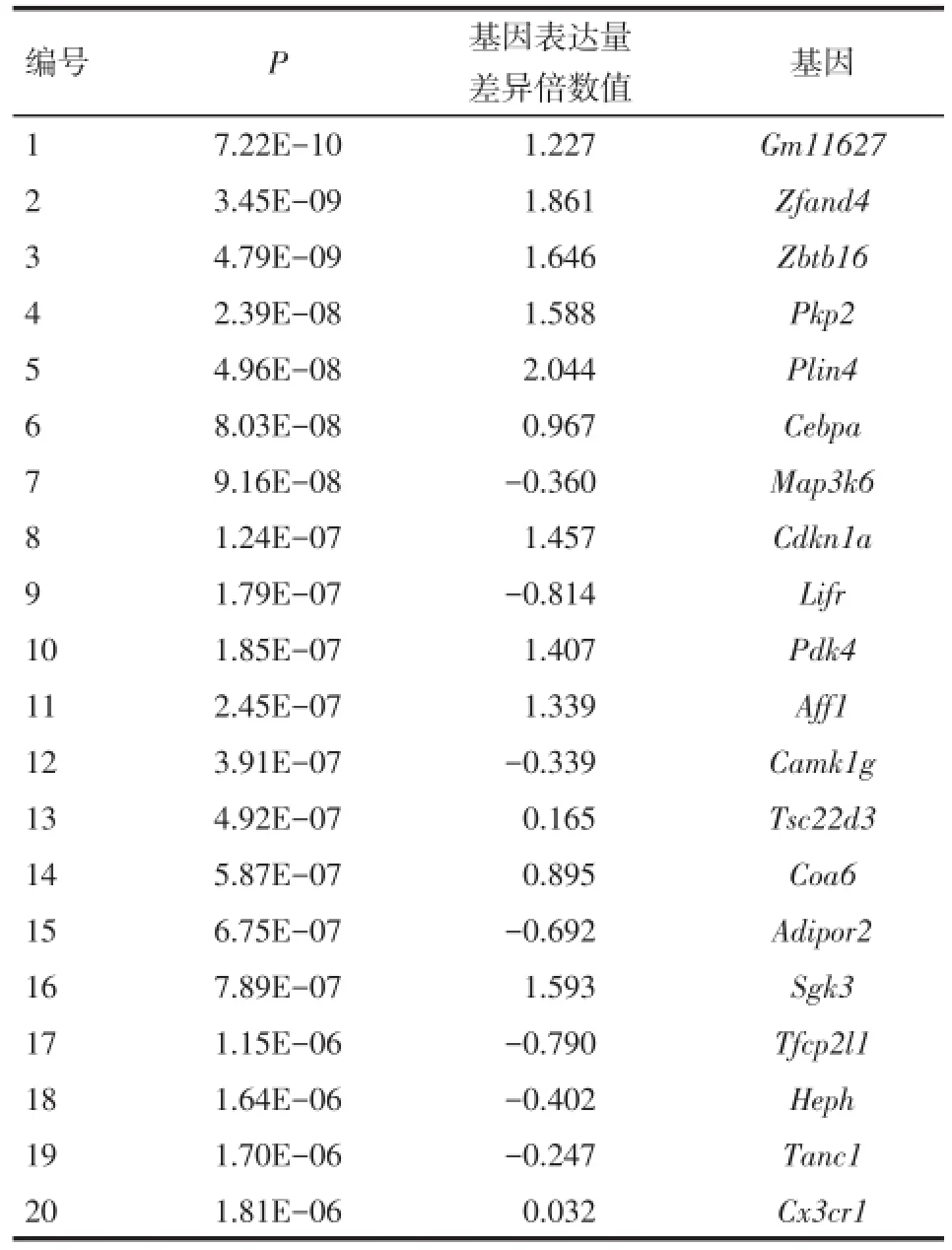
Tab.1 Top 20 differentially expressed genes identified from the microarray datasets of E-GEOD-7762表1 前20个E-GEOD-7762微阵列数据识别的差异表达基因
3 讨论
Korostynski等[2]对小鼠纹状体转录组进行分析,研究吗啡急性处理后4只不同种属的小鼠之间转录应答的区别,以及吗啡慢性处理后的基因表达情况。本文对吗啡急性处理小鼠的微阵列数据进行质量控制分析,识别其差异表达基因,并进行差异表达基因的通路富集分析。质量控制报告显示芯片数据均一性和阵列强度相似性良好。本文共识别到Gm11627、Zfand4、Zbtb16、Pkp2和Plin4等481个差异表达基因。表达差异最为显著的前20个基因在吗啡处理后的表达量变化显著,差异表达基因富集于癌症通路、黑素原生成和丝裂原活化蛋白激酶信号通路等8条代谢通路之中,且差异表达基因的富集程度较高,通路最小基因数均≥5,具有统计学意义。

Tab.2 Pathway enrichment analysis based on KEGG表2 基于KEGG的通路富集分析
本文所识别的Gm11627、Zfand4、Zbtb16、Pkp2 和Plin4等差异表达基因在吗啡对小鼠急性处理前后显著差异表达,并与该过程导致的生理变化有一定关系。已有研究证明,uGTZB7基因序列的多态性影响吗啡的体内代谢[15]。大鼠JNK3和GRK基因的表达分别与脑内神经细胞损伤和吗啡成瘾,以及吗啡急性处理过程中的耐受有关[16]。差异表达基因Zbtb16作为一种转录抑制剂,与吗啡成瘾现象和小鼠脑纹状体的细胞分化功能密切相关[17]。此外,Pkp2基因是导致心律失常性右心室心肌病的重要致病基因[18]。本研究KEGG通路富集分析结果显示,包括Pkp2在内的6个差异表达基因富集在致心律失常性右心室心肌病代谢通路中,进一步印证了Pkp2在心肌病表达方面的功能,提示吗啡急性处理后Zbtb16、Pkp2等基因的差异表达可能影响细胞分化,并可能导致吗啡成瘾和心肌病。
已有研究显示,丝裂原活化蛋白激酶信号广泛存在于神经元中[19],通过影响神经突触的可塑性产生对吗啡的病理性镇痛耐受作用[20]。丝裂原活化蛋白激酶信号通路在前额叶皮质磷酸化过程中导致细胞外调节蛋白激酶表达降低,进而参与吗啡依赖的形成和戒断反应,造成吗啡成瘾[21]。此外,丝裂原活化蛋白激酶信号通路具有信号转导作用,因此其在细胞的增殖、分化及凋亡等生化反应过程中起到重要作用。而鞘脂类在脂筏的形成过程中扮演着重要角色,其代谢过程与细胞的信号转导有关[22]。同时,焦点黏附通常在焦点黏附激酶的作用下发生,该过程可以间接反映细胞与细胞外基质之间的信号传递,与细胞迁移、侵袭、存活等生物过程密切相关[23]。代谢通路的富集反映出,吗啡急性处理后可能发生丝裂原活化蛋白激酶信号转导、鞘脂的代谢和焦点黏附等生理过程。
综上所述,吗啡急性处理后可能发生细胞分化、丝裂原活化蛋白激酶信号转导、鞘脂的代谢和焦点黏附等生理过程,并可能导致成瘾和心肌病。上述差异表达基因及其富集的代谢通路可成为吗啡对小鼠急性处理潜在的生物标记,为进一步研究吗啡对人体的作用机制提供了依据。目前生物信息学分析主要集中在多元数据网络的分析及可视化,蛋白质相互作用的评估和预测,网络聚类分析和相关子网络的构建,以及生物功能和代谢通路分析等方面[24]。因此,微阵列数据的扩展、差异表达基因的蛋白质相互作用网络分析、基因本体分析等,都可能成为今后关于吗啡对人体作用机制的研究方向。
(图3、5见插页)
[1]赵倩华.吗啡给药途径及其新剂型的研究进展[J].医药论坛杂志,2013,34(1):143-145.
[2]Korostynski M,Piechota M,Kaminska D,et al.Morphine effects on striatal transcriptome in mice[J].Genome Biol,2007,8(6):R128.
[3]徐娟.miRNA-miRNA协同调控网络:构建、疾病miRNA拓扑特征及序列和结构相似性分析[D].哈尔滨:哈尔滨医科大学,2011.
[4]Li C,Shen W,Shen S,et al.Gene expression patterns combined with bioinformatics analysis identify genes associated with cholan⁃giocarcinoma[J].Comput Biol Chem,2013,47:192-197.
[5]Gautier L,Cope L,Bolstad BM,et al.Affy--analysis of Affymetrix GeneChip data at the probe level[J].Bioinformatics,2004,20(3): 307-315.
[6]Wilson CL,Miller CJ.Simpleaffy:a BioConductor package for Af⁃fymetrix Quality Control and data analysis[J].Bioinformatics,2005,21(18):3683-3685.
[7]Raman T,O'Connor TP,Hackett NR,et al.Quality control in micro⁃array assessment of gene expression in human airway epithelium[J]. BMC Genomics,2009,10(1):493.
[8]DUNDAS JB.Affymetrix Quality control[EB/OL].(2011-3-7)[2014-07-26]http://bioinformatics.gr/tutorials/microarray-technol⁃ogy-basics/maqc/95-affyqc.html.
[9]Kim Y,Doan BQ,Duggal P,et al.Normalization of microarray ex⁃pression data using within-pedigree pool and its effect on linkage analysis[J].BMC proceedings BioMed Central Ltd,2007,1(Suppl 1):S152.
[10]Smyth GK.Linear models and empirical bayes methods for assess⁃ing differential expression in microarray experiments[J].Stat Appl Genet Mol Biol,2004,3(1):3.
[11]Reiner A,Yekutieli D,Benjamini Y.Identifying differentially ex⁃pressed genes using false discovery rate controlling procedures[J]. Bioinformatics,2003,19(3):368-375.
[12]Huang DW,Sherman BT,Lempicki RA.Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources [J].Nature protocols,2009,4(1):44-57.
[13]Ford G,Xu Z,Gates A,et al.Expression Analysis Systematic Ex⁃plorer(EASE)analysis reveals differential gene expression in per⁃manent and transient focal stroke rat models[J].Brain Res,2006,1071(1):226-236.
[14]Huang DW,Sherman BT,Lempicki RA.Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources [J].Nat Protoc,2009,4:44-57.
[15]龚妍,曹迪,李智平.UGT2B7代谢酶,阿片μ受体及ABCB1基因多态性对小儿吗啡镇痛效能影响的研究进展.第二十三届全国儿科药学学术会议论文集[C].石家庄:中国药房杂志社,2012.
[16]范学良.吗啡对GRKZ、GRKS和JNK3基因在大鼠脑内表达调控的研究[D].上海:复旦大学,2002.
[17]Piechota M,Korostynski M,Solecki W,et al.The dissection of tran⁃scriptional modules regulated by various drugs of abuse in the mouse striatum[J].Genome Biol,2010,11(5):R48.
[18]张明昌,张宁生,童奇,等.PKP2基因突变与青壮年猝死综合征.全国第九次法医学术交流会论文集[C].北京:中国法医学会,2013.
[19]Shen N,Guo RX,Hu F,et al.Spinal Cx43 mediates chronic mor⁃phine antinociceptive tolerance through JNK pathway in rats[J].CHI⁃NESE PHARMACOLOGICAL BULLETIN,2013,29(03)372-376.
[20]张乐,朱永平.丝裂原活化蛋白激酶信号通路在吗啡依赖中的作用[J].中国药物依赖性杂志,2012,21(2):100-104.
[21]阎春霞,李阳,张宏星,等.脑前额叶皮层MAPK通路在吗啡成瘾记忆中的作用.全国第九次法医学术交流会论文集[C].北京:中国法医学会,2013.
[22]刘圣红,苟萍.鞘脂类的研究进展[J].生物技术,2009,19(2):96-98.
[23]袁周:焦点粘附激酶在肝癌中表达及其对人肝癌细胞MHCC97-H作用的研究[D].上海:中山医院,2006.
[24]陈刚.生物网络分析及其在复杂疾病研究中的应用[D].长沙:中南大学,2012.
(2014-07-28收稿2014-08-15修回)
(本文编辑陆荣展)
Microarray datasets and biomarker in mice who were acutely administered with morphine
ZHANG Wenhua1,WANG Yanping2
1 Department of Anesthesiology,Third Hospital Affiliated to Qiqihaer Medical College,Qiqihaer,Heilongjiang 161000,China;2 Evidence-Based Medicine Technology Development Center in Jinan
△Corresponding AuthorE-mail:1102207555@qq.com
ObjectiveTo identify the differentially expressed genes and functional enrichment pathways using bioin⁃formatics technology in mice who were acutely administered with Morphine.MethodsFirst,we downloaded microarray da⁃tasets of mice which were acutely administered with Morphine from Gene Expression Omnibus,and assess the quality control parameters of the microarray datasets using AffyQCReport 1.42.0 package by R programming language 3.1.0 software.Subse⁃quently,the differentially expressed genes of the microarray datasets were identified using the linear models for microarray data of R language.Finally,functional pathways of the differentially expressed genes were enriched based on expression anal⁃ysis and systematic explorer.ResultsThe microarray datasets showed preferable uniformity and intension similarity.A to⁃tal of 481 differentially expressed genes including Gm11627,Zfand4,Zbtb16,Pkp2 and Plin4 were identified.While 8 func⁃tional enrichment pathways,including pathways in cancer,melanogenesis and mitogen-activated protein kinase signaling were revealed.ConclusionThe differentially expressed genes and functional pathways were the underlying biomarkers of mice who were acutely administered with Morphine.
morphine;mice;microarray analysis;gene expression;biological markers;enrichment of functional analysis
R318.04
ADOI:10.11958/j.issn.0253-9896.2015.02.013
2012年国家医学教育发展中心课题项目(2012-03-07-142)
1黑龙江齐齐哈尔,齐齐哈尔医学院附属第三医院麻醉科(邮编161000);2济南,济南循证医药科技开发中心科研
张文华(1972),女,硕士,副主任医师,主要从事麻醉技术及麻醉用药研究
△通迅作者E-mail:1102207555@qq.com
猜你喜欢
介入放射学杂志(2022年10期)2022-11-02
科技创新与应用(2021年7期)2021-02-04
医药前沿(2020年23期)2020-12-03
科学(2020年2期)2020-08-24
中成药(2018年10期)2018-10-26
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年3期)2015-06-10
医学研究杂志(2015年3期)2015-06-10
中国医药导报(2015年24期)2015-02-28
创业家(2015年1期)2015-02-27
