
生物学软件在核酸序列比对与系统发育分析中的应用
2015-08-17 17:47王小国
现代农业科技 2015年12期
关键词:构建
王小国

摘要 通过ClustalX2.1、Boxshade、DNAman和MEGA6等软件的使用,介绍了生物学软件在核酸序列比对、着色美化、序列分析和植物系统发育树构建中的应用。并结合具体实例,采用邻接法对核基因组中的内转录间隔区序列进行了构树,进化枝的可信度较高,表明该方法适用于相似度较高、亲缘关系较近序列的系统发育树的构建。
关键词 序列比对;邻接法;系统发育树;构建
中图分类号 Q7;TP31 文献标识码 A 文章编号 1007-5739(2015)12-0347-02
Application of Biology Software in the Nucleic Acid Sequence Alignment and Phylogenetic Analysis
WANG Xiao-guo
(Sanmenxia Polytechnic,Sanmenxia Henan 472000)
Abstract This paper introduced the application of biology software in the nucleic acid sequence alignment,beautification,sequence analysis and plant phylogenetic tree construction through the use of software such as ClustalX 2.1,Boxshade,DNAman and MEGA 6.Combined with the specific example,constructed neighbor-joined tree according to internal transcribed spacer of nuclear genome sequences,and the credibility of clade was higher,so the method was suitable for the close genetic relationship sequence in the construction of phylogenetic tree.
Key words sequence alignment;neighbor-joined method;phylogenetic tree;construct
核酸序列比对(Nucleic acid sequence alignment)又称为核酸序列联配,是通过2个或多个核酸序列的比对,显示出其中相似结构区域的过程[1],是进一步进行相似性、序列间亲缘关系和系统发育分析的基础[2-3]。在系统发育分析中,最常用的可视化表示进化关系的方法就是绘制系统发育进化树(Phylogenetic trees)。系统发育树构建有非加权分组平均法(Unweighted-pair-groop-method with arithmetic means,UPGMA)、邻接法(Neighbor-joined,NJ)、最大简约法(Maximum parsimony,MP)和最大似然法(Maximum likelihood,ML)等多种方法[4],由于邻接法具有构建的树相对准确、假设少且计算速度快的特点,因而是目前最常用的方法之一。但NJ法所分析序列的遗传距离不能太大,适用于相似度较高、亲缘关系较近的短序列[5]。
核基因组序列中的内转录间隔区(Internal transcribed spacer,ITS)主要编码植物的核糖体RNA,属于中度保守序列,因此可用于较低分类阶元的系统发育研究[6-7]。本文以Ziziphus Mill.的部分ITS序列为对象,介绍了ClustalX2.1[8-9]、Boxshade、DNAman和MEGA6[10]等软件在序列比对、着色美化、序列分析和植物系统发育树构建中的应用。
1 序列获取
从NCBI网站(http://www.ncbi.nlm.nih.gov/)下载Ziziphus Mill.的4条ITS核酸序列,以Fasta格式存储于桌面文档中,命名为four sequences.txt。
2 使用ClustalX进行多序列比对
2.1 打开ClustalX软件
找开ClustalX软件(Version 2.1,2010年发布,http://www.clustal.org/),在“Multiple alignment”模式下点击“File”菜单→“Load sequece”,打开存储于桌面上的four sequences.txt文件(图1)。
2.2 进行序列比对
打开“Alignment”菜单→“Do complete alignment”,系统提示“Do complete alignment”结果的输出文件类型及保存位置(图2)。其中,dnd格式为输出向导树文件,可以用Treeview软件打开,aln为输出比对文件格式,打开后对序列进行手动调整。
2.3 aln文件的着色美化
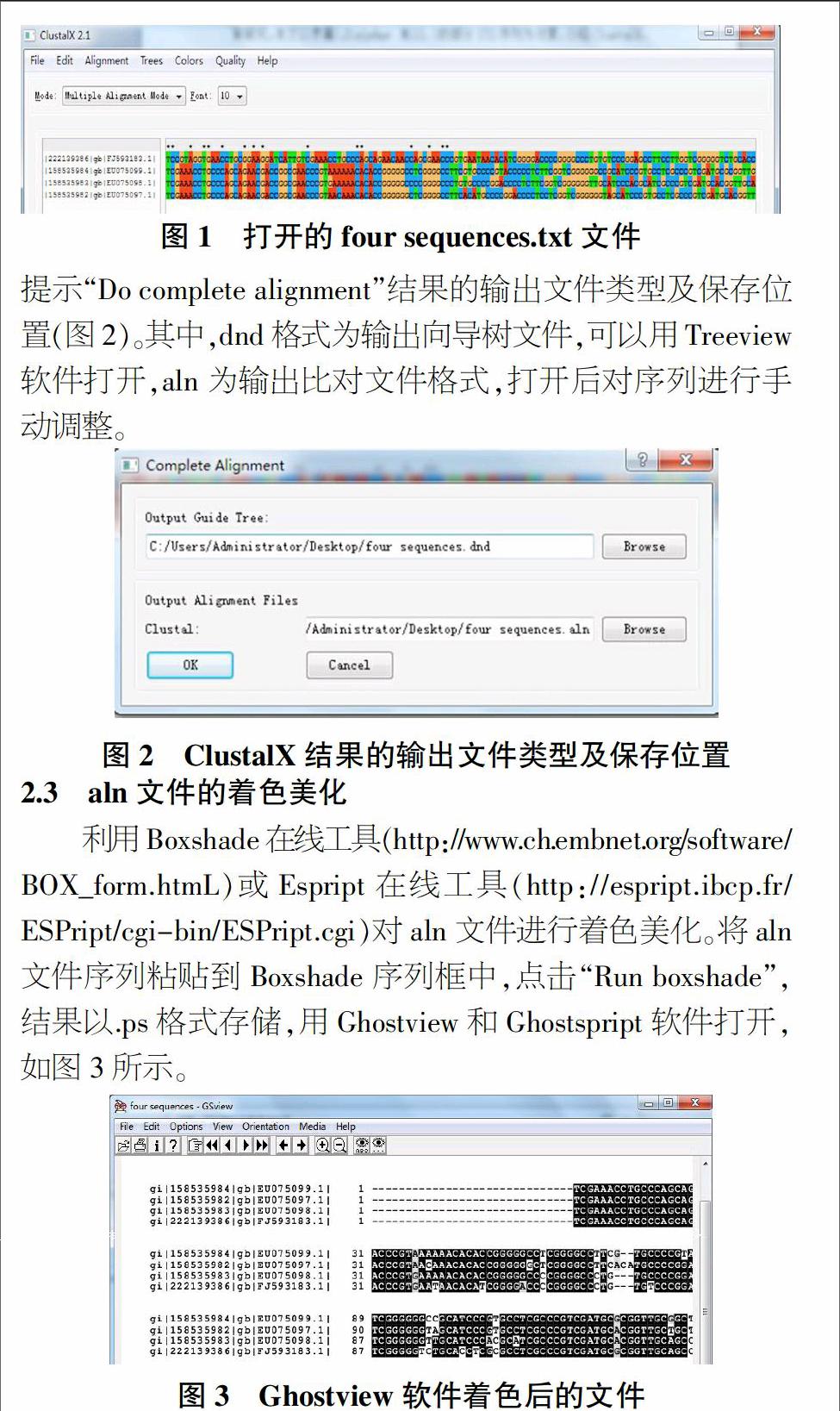
利用Boxshade在线工具(http://www.ch.embnet.org/software/BOX_form.htmL)或Espript在线工具(http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi)对aln文件进行着色美化。将aln文件序列粘贴到Boxshade序列框中,点击“Run boxshade”,结果以.ps格式存储,用Ghostview和Ghostspript软件打开,如图3所示。
3 DNAman多序列比对和序列一致性endprint
DNAman软件除了可对序列进行比对之外,还可进行引物的设计、限制性内切酶和质粒图谱的绘制,功能较为强大。双击DNAman图标,打开“File”菜单→“Open”,选择four sequences.aln文件,再打开“Sequence”菜单→“Alignment”→“Multiple sequence alignment”,从显示结果可看出,这4条ITS序列的序列一致性为90.10%(图4),结果可以以Phylip、Clustal和GCG等多种格式输出。
4 MEGA进行系统发育分析
4.1 打开分子进化遗传分析(Molecular evolution genetic analysis,MEGA)软件
由于MEGA只能识别.meg文件格式,故需要对.aln格式的文件进行转化。点击“File”→“Convert file format to MEGA”,将.aln格式文件转化为.meg格式,存储于桌面上,命名为four sequences.meg。或进行在线文件格式转换(http://sing.ei.uvigo.es/ALTER/)。
4.2 主窗口中打开“Phylogeny”菜单
可以看到,MEGA提供了UPGMA法、NJ法、MP法和ML法等多种算法,如前所述,NJ法在处理相似度较高、亲缘关系较近的序列时是最可靠的一种算法。选择“Construct/Test neighbor-joined tree”,在弹出的对话框“Options summary”→“Test phylogeny”中,选择“Bootstrap method”,重复次数为1000次,模式框中选择Kimura-2-parameter model对进化树进行评估(图5),本次所构建的系统进化树如图6所示。
从构建的NJ树可知,基因登录号为EU075099和EU07 5097的序列聚为一类,bootstrap支持率为97%,表明该进化枝的可信度较高。
5 结语
本文以Ziziphus Mill.的部分ITS序列为对象,介绍了ClustalX、Boxshade、DNAman和MEGA等软件在序列比对、着色美化、序列分析和植物系统发育树的构建中的应用。在此需要特别指出,在基于序列构建进化树的过程中,由于不同的算法,不同的重复次数,选用不同的模型所构建的树是不完全相同的[11],因此,这种单纯的依靠某段核酸序列建立的进化树只能提供物种进化的部分信息,而不能完全代表该物种进化的全过程。
6 参考文献
[1] 王禄山,高培基.生物信息学应用技术[M].北京:化学工业出版社,2007.
[2] 谢强,卜文俊.核苷酸序列比对在生物系统发育研究中的应用[J].动物分类学报,2005(2):281-286.
[3] 杨学森.基于汉明距离的DNA短序列比对算法研究[D].哈尔滨:哈尔滨工业大学,2013.
[4] 路明.利用进化树研究基因序列的进化[D].南宁:广西大学,2014.
[5] 高凯.NJ进化树构建方法的改进及其应用[D].北京:北京工业大学,2008.
[6] 樊杰,白妍,束明月.远志属7种药用植物ITS1和ITS2序列分析[J].中草药,2015(4):562-565.
[7] BRUCE G,BALDWIN.Phylogenetic utility of the internal transcribed spacers of nuclear ribosomal DNA in plants:An example from the compositae[J].Molecular Phylogenetics and Evolution,1992,1(1):3-16.
[8] LARKIN M A,BLACKSHIELDS G,BROWN N P,et al.Clustal W and Clustal X version 2.0[J].Bioinformatics,2007,23:2947-2948.
[9] 李彬彬,黄培春,钟复光.生物学软件在线粒体DNA序列多态性分析中的应用[J].生物信息学,2010(2):153-155.
[10] TAMURA K,STECHER G,PETERSON D,et al.MEGA6:Molecular Evolutionary Genetics Analysis Version 6.0[J].Molecular Biology and Evolution,2013,30:2725-2729.
[11] 李衍达,孙之荣译.生物信息学[M].北京:清华大学出版社,2008.endprint
猜你喜欢
企业导报(2016年19期)2016-11-05
企业导报(2016年19期)2016-11-05
时代金融(2016年23期)2016-10-31
科技视界(2016年22期)2016-10-18
科学与财富(2016年28期)2016-10-14
科学与财富(2016年28期)2016-10-14
大学教育(2016年9期)2016-10-09
成才之路(2016年26期)2016-10-08
成才之路(2016年25期)2016-10-08
