
超高效液相色谱-三重四极杆质谱联用同时检测环境水体中22种典型药品及个人护理用品
2015-08-03 09:26:48吴春英陆文龙
色谱 2015年8期
吴春英, 谷 风, 白 鹭, 陆文龙
(1.吉林化工学院资源与环境工程学院,吉林吉林132022;2.清华大学环境学院,环境模拟与污染控制国家重点联合实验室,北京100084)
药品和个人护理用品(pharmaceuticals and personal care products,PPCPs)由于其潜在的环境风险近年来备受关注,PPCPs包括各类化学物质,例如各种处方药和非处方药(如抗生素、类固醇、消炎药、镇静剂、抗癫痫药、止痛药、降压药等)、香料、化妆品等[1-3]。大多数PPCPs是水溶性的,并且在环境中具有累积性、持久性、生物毒性和“三致”效应等,对生物体的影响不可逆转;随着生产量和使用量的逐年增加,对环境的危害也越来越显著[1,4-6],其引起的潜在的环境风险越来越引起广泛关注。因此,建立一种快速、灵敏、可靠的PPCPs定量检测方法是开展相关研究工作的重要基础。经过数年的研究工作,形成了多种分析方法[5,6],基本上是固相萃取等前处理方法和色谱或色谱-质谱联用,尽管其中色谱-质谱联用法具有检出限低、灵敏度高、线性范围宽等优点,但由于药品种类繁多,性质、分子结构各有差异,分析方法的传统复杂性使得监测工作浪费了大量的人力物力[7-10]。
本研究参考文献[7,10-13],根据国内 PPCPs 的生产和使用情况及在环境中的检出水平[5,12-15],选取了包括抗生素、消炎止痛、抗癫痫、调血脂、神经类、强心剂、支气管扩张剂、β-受体阻滞剂和广谱蚊虫驱避剂等11类22种典型的PPCPs作为研究对象,采用固相萃取及UPLC-MS/MS法对环境水同时进行预处理和分析,并对萃取柱、淋洗液、色谱条件等进行优化,在保证高回收率的前提下,降低目标物的检出限,统一预处理方法,极大地缩短预处理时间,有效降低预处理成本。该法可应用于实际样品的检测,为PPCPs的相关研究提供可靠的分析和检测方法。
1 实验部分
1.1 试剂
实验所用22种目标物(见表1)标准品及内标沙丁胺醇-氘 3(salbutamol-d3)购自 Dr.Ehrenstorfer(Germany),各标准品的纯度均在98.5%以上。以甲醇为溶剂配制1 g/L的各标准品储备液,置于-20℃保存。甲醇、甲酸、二氯甲烷、乙腈等购自Fisher(USA),实验用所有溶剂均为HPLC纯或更高纯度。维生素C(Vc)、Na2EDTA均购自Sigma-Aldrich(USA)。试验所用水均为超纯水(ultra pure water,UPW)。
1.2 仪器与设备
选用带自动进样器的ACQUITY UPLC系统(Waters,USA)和Finnigan TSQ Quantum Discovery MAX三重四极杆质谱分析仪;12通道半自动反相固相萃取装置(Supelco,USA);Oasis HLB SPE 小柱(6 mL/200 mg)和净化用 Sep-Pak Plus C18小柱(3 mL/500 mg)购自 Waters,USA;NEVAP12氮吹仪(Organomation,USA);CF16RXII型离心机(日本);Milli-Q超纯水器(美国Millipore公司)。所用的玻璃纤维滤膜GF/B(1 μm)由英国Whatman提供。

表1 目标物的MS/MS分析条件Table 1 MS/MS parameters for the targets
1.3 样品的预处理
水样中加入维生素C,使其质量浓度为1 g/L;用稀盐酸将样品的pH值调至3,经玻璃纤维滤膜(GF/B)过滤,在透过液中加入 Na2EDTA,质量浓度为1 g/L,用以屏蔽样品中的金属离子。用3×3 mL甲醇及3×3 mL UPW活化SPE小柱,然后上样进行固相萃取,萃取流速为10 mL/min。萃取结束后,用真空泵继续抽吸1 h。SPE小柱串接Plus C18净化小柱进行洗脱,以甲醇(2×3 mL)为洗脱剂,将洗脱液收集于10 mL棕色玻璃试管中。在洗脱液中加入100 ng/L内标沙丁胺醇-d3用来定量,然后在柔和氮气下吹干,再用1 mL甲醇/水(70∶30,v/v)溶解。复溶后离心(离心参数:4℃,3 000 r/min,15 min),取上清液进行 UPLC/MS/MS分析。
1.4 UPLC与MS条件
UPLC条件 色谱柱为反相ACQUITY UPLCTMBEH C18 柱 (100 mm × 2.1 mm,1.7 μm;Waters)。流动相为含0.1%甲酸的水溶液-甲醇(7∶3,v/v)。流速为 0.35 mL/min,无分流。柱温为50℃,进样量为10 μL。由于MS是采用多反应监测(MRM)模式,检测并不需要目标物间完全分离。在下一次进样前,色谱柱先平衡9 min。
质谱条件 ESI源,高纯氮气用作脱溶剂和雾化,采用氩气(99.998%)作碰撞气。脱溶剂时温度350 ℃,流速 550 mL/min,锥孔气流速 70 mL/min。源温设置为120℃以防溶剂再凝结。雾化器压力为310 kPa(45 psi),检测方式为 MRM。
2 结果与讨论
2.1 色谱条件的选择
每一种目标物在不同色谱柱上的保留时间不同,使用反相ACQUITY UPLCTMBEH C18分离的峰形尖锐对称,且具有相同母离子及碎片离子的目标物能够完全分离,信噪比较高。因此本文选择该色谱柱作为分离柱。
使用 UPLC-MS/MS 检测,对比了文献[11-15]中报道较多的甲醇、乙腈及其和水不同比例的混合溶液作为流动相的结果。当选用乙腈作为流动相时,目标物均得到了很好的分离和检测结果,但双氯芬酸的响应强度较小,氯贝酸和苯氧苯丙酸均未检出;选用甲醇作流动相时,氯贝酸和苯氧苯丙酸虽然都能检出,但响应强度非常弱,而且个别目标物分离效果不理想;选含0.1%甲酸的水-甲醇(7∶3,体积比,下同)为流动相时,响应强度相对较小的双氯芬酸、氯贝酸和苯氧苯丙酸在200 μg/L时均获得较好的响应值(5.52×103、0.96 ×104和1.84 ×104),明显优于甲醇作为流动相时的分离效果,且所有目标物的分离效果非常好。因此,本研究选择含0.1%甲酸的水-甲醇(7∶3)作为流动相。
2.2 质谱的优化
MS系统采用QuanOptimize方式进行自动优化,用甲醇将22种目标物配成混合标准溶液,在全扫描模式下扫描,找出准确的[M+H]+分子离子峰,然后对其进行子离子扫描以获得二次碎裂产生的子离子。以分子离子和2个响应适中的子离子组成检测离子对,以MRM模式进行检测,在此基础上重点优化对灵敏度影响较大的碎裂电压和碰撞能量,使选定的母离子和子离子组成的特征离子对的丰度和比例达到最佳。优化每一目标物的MS/MS参数,包括电离模式、母离子、子离子、锥孔电压(CV)、碰撞能(CE),得到的主要分析参数如表1所示。数据采集采用MRM模式,驻留时间为0.1 s,通道滞迟时间为0.2 s。对于每一电离模式,采用离子响应值最弱的目标物对系统进行调谐。
2.3 固相萃取条件的选择与优化
为提高预处理效果,保证分析的准确性和灵敏性,在目标物的萃取过程中,对SPE小柱和淋洗溶剂进行了对比选择,以标准品的回收率为评价指标[15-17]考察了两种小柱(Sep-Pak Plus C18和Oasis HLB),3种淋洗液(甲醇、二氯甲烷和乙腈)。结果表明,当固相萃取小柱为Oasis HLB、淋洗液为甲醇时,各目标物的回收率均在83%(美托洛尔)~110%(咖啡因)之间,RSD最大的DEET也只有17.2%。因此,本试验采用固相萃取柱Oasis HLB,淋洗液为甲醇。
2.4 基质效应评价
样品中的内源性组分和样品处理过程中引入的杂质对目标物检测结果的影响称为基质效应,它可影响线性关系、检出限(LOD)、定量限(LOQ)等测定指标。本研究采用标准曲线法考察了水样对PPCPs目标物检测的基质效应。结果表明,基质校准曲线的斜率比纯溶剂标准曲线的斜率小,即存在基质抑制效应。在水样中加入内标物对待测组分的测定无任何干扰,且受基质成分的影响也极为相似。内标物可抵消质谱离子化时的基质效应。因此,能最大限度地消除基质效应干扰,本研究采用基质匹配标准曲线和内标法进行定量,可得到目标物的准确测定结果。
2.5 线性范围与检出限
样品中目标物的定量通过与内标沙丁胺醇-氘3的比较来实现,该内标在ESI阳离子模式下响应效果很好,但在负离子模式下响应效果较差。内标在阳离子模式下获得的峰值也用于双氯酚酸、氯贝酸和苯氧苯丙酸这3种在负离子模式下检测的物质定量。各目标物标准品在UPLC-MS/MS系统中有效的线性响应(R2>0.997)浓度范围如表2所示,该范围与目标物有关。校准采用2~2 000 μg/L(10点)混合标准溶液进行,混合标准溶液中含有100 ng/L内标。分别以各目标物色谱峰的信噪比S/N=3和S/N=10确定检出限和定量限,结合所取样品体积及最终定容体积,可得水中22种PPCPs的检出限和定量限(见表2)。与以往的分析方法相比,该法具有较宽的线性范围和较低的检出限和定量限[13,15],可满足环境样品中目标物的分析。
2.6 回收率与精密度
在不同基质中加入目标物的标准品以确定该基质中目标物的回收率。基质包括UPW和某污水处理厂(采用厌氧/缺氧/好氧-膜生物反应器(A2/OMBR)工艺)的进、出水。水样中加入维生素C,使其质量浓度为1 g/L,目的是防止各药品成分分解[13]。向300 mL的 UPW、WWTP 进水、WWTP 出水中分别加入0.3、0.6和0.3 mL的标准品溶液(1 mg/L)。所有的回收率试验均做4份平行样品,并考虑扣减过程空白。目标物的回收率均为73%~125%,RSDs不大于18%(见表3)。
2.7 实际样品的测定
为了验证本方法在实际水样测定中的有效性,对自来水厂及两个污水处理厂的出水、松花江水和校园生活污水中的22种PPCPs含量进行实际检测;为确保定量准确,在每批次样品分析中进行加标回收率测定,测定的浓度按回收率进行校正;此外,在每批样品测定时,过程空白与溶剂空白也同时进行处理和分析。实验结果如表4所示,可以看出,实际样品中均能检出双氯芬酸、氯贝酸和咖啡因,且含量在所检出的PPCPs中较高,说明处理工艺对这3种PPCPs的去除仍有一定的局限性,在日常生活的使用管理和水处理中对该类PPCPs应给予更多关注。

表2 目标物的线性关系、LODs和LOQsTable 2 Linear relationships,LODs and LOQs of the targets
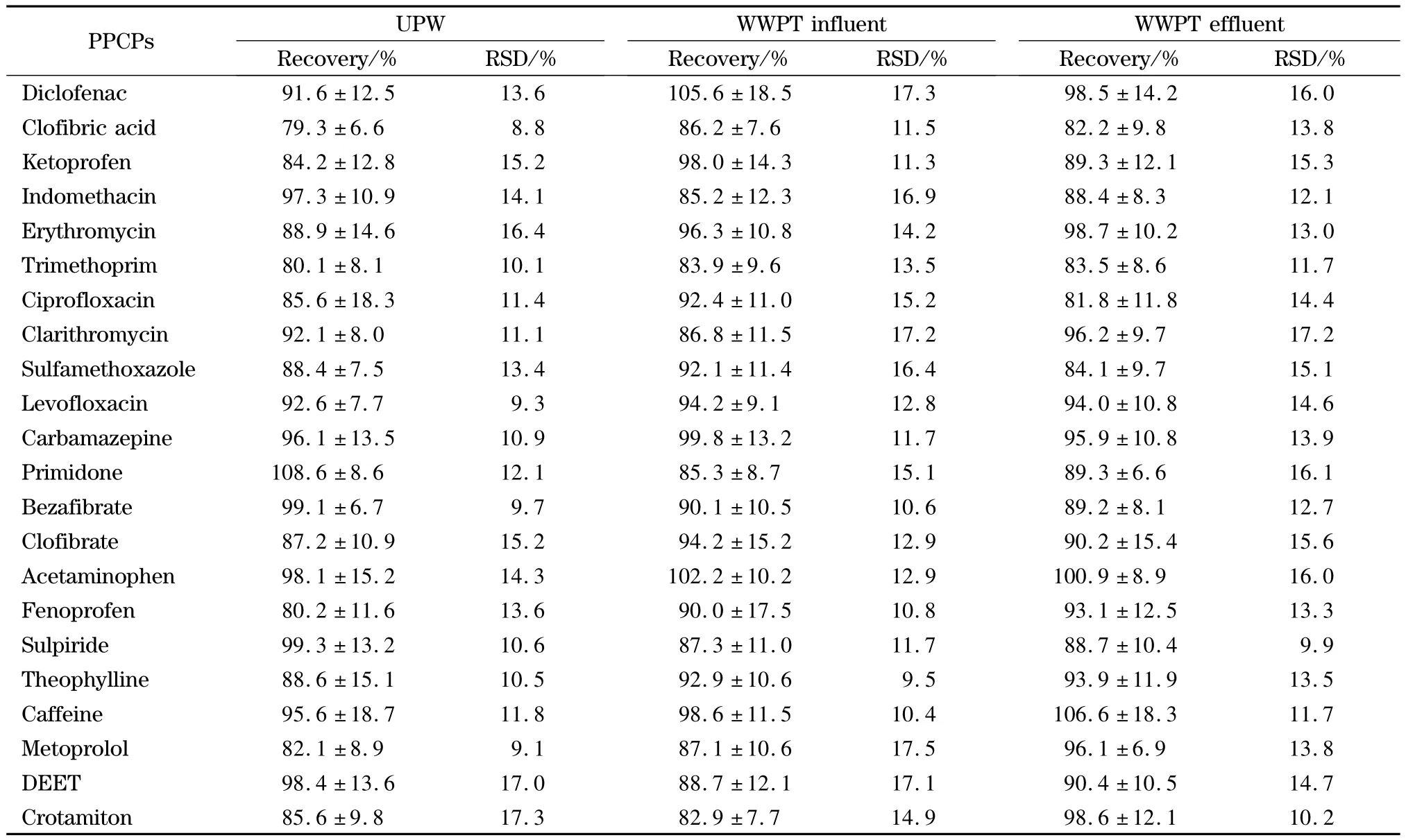
表3 不同基质中目标物的回收率和精密度(n=4)Table 3 Recoveries and precisions(REDs)of targets in different matrices(n=4)

表4 实际水样中PPCPs的含量Table 4 Contents of PPCPs in actual water samples ng/L
3 结论
采用固相萃取、超高效液相色谱-串联质谱建立了环境水中22种典型PPCPs的分析方法。该方法具有检出限低、回收率高、线性关系良好等优点,适用于多种目标物同时检测,减少时间和材料的能耗,为其在环境中的迁移转化及去除的相关研究提供支持和帮助。
[1] EPA.Protecting Human Health,Safeguarding the Natural Environment.(1999-12-24)[2005-03-20].http:/www.epa.gov/nerl/research/1999 /html/g8-14.html
[2] Zhao X,Chen Z L,Wang X C,et al.Bioresource Technol,2015,179:104
[3] Carballa M,Omil F,Lema J M,et al.Water Res,2004,38(12):2918
[4] Huang C L,Ma H W,Yu C P.Sci Total Environ,2014,499(15):265
[5] Jia Y Y,Tan J H,Xu C,et al.Chinese Journal of Chromatography(贾妍艳,谭建华,徐晨,等.色谱),2014,32(3):263
[6] Zhu S C,Wang J,Shao W W,et al.Chinese Journal of Chromatography(朱赛嫦,王静,邵卫伟,等.色谱),2013,31(1):15
[7] Kong L X,Kadokami K,Wang S P,et al.Chemosphere,2015,122:125
[8] Blair B D,Crago J P,Hedman C J,et al.Chemosphere,2013,93(9):2116
[9] Kasprzyk-Hordern B,Dinsdale R M,Guwy A J.J Chromatogr A,2007,1161(1/2):132
[10] Cerqueira M B R,Guilherme J R,Caldas S S,et al.Chemosphere,2014,107:74
[11] Kuroda K,Nakada N,Hanamoto S,et al.Sci Total Environ,2015,506/507:287
[12] Jia X X.[MS Dissertation].Xi’an:Xi’an University of Architecture and Technology(贾翔宵.[硕士学位论文].西安:西安建筑科技大学),2010
[13] Zhou H D.[Postdoctoral Research Report].Beijing:Tsinghua University(周海东.[博士后研究报告].北京:清华大学),2009
[14] Yan Z,Nie J Y,Xu G F,et al.Journal of Instrumental Analysis(闫震,聂继云,徐国锋,等.分析测试学报),2014,33(9):1000
[15] Zhang P W,Zhao G F,Zhou H D,et al.Environmental Monitoring of China(张盼伟,赵高峰,周怀东,等.中国环境监测),2014,30(1):138
[16] Gao H,Zu G R,Gu J,et al.Chinese Journal of Analysis Laboratory(高会,祖国仁,顾佳,等.分析试验室),2011,30(12):8
[17] Qiu Y Q,Chen G Q,Lei Y,et al.Physical Testing and Chemical Analysis Part B:Chemical Analysis(邱蕴绮,陈国权,雷毅,等.理化检验-化学分册),2014,50(6):57
猜你喜欢
化工设计通讯(2022年10期)2022-12-31 20:42:50
少年文艺(2022年8期)2022-07-08 10:02:47
波谱学杂志(2022年2期)2022-06-14 09:52:02
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
中国特种设备安全(2021年12期)2021-04-26 14:37:00
中成药(2018年6期)2018-07-11 03:01:32
中国经济周刊(2017年6期)2017-03-21 00:59:27
读写算·高年级(2016年3期)2016-05-30 01:53:46
中国粮油学报(2016年5期)2016-01-23 02:45:06
中国医疗美容(2015年1期)2015-07-12 10:06:18
