
超高效液相色谱-串联质谱法测定调制乳中3种磺胺类药物残留
2015-08-03 09:27:00程国栋吴小慧仝面换高建军
色谱 2015年8期
程国栋, 吴小慧, 金 珠, 张 宇, 郝 单, 仝面换, 高建军
(蒙牛乳业(马鞍山)有限公司,安徽 马鞍山243000)
磺胺类药物(SAs)是对氨基苯磺酰胺结构药物的总称,是一类用于预防和治疗细菌感染性疾病的抗菌药,因其抗菌谱广和价廉、易得等特点,一直在兽医临床和畜牧业中广泛应用[1]。因残留有抗生素的畜产品中被人类长期食用后会对人类健康产生巨大危害,中国、欧盟、美国食品药品监督管理局(FDA)以及日本等许多国家和监管机构都已经对该类抗生素在牛奶中的残留限量做了明确规定[2]。目前磺胺类药物的检测方法主要有酶联免疫法[3]、高效液相色谱法[4-8]、胶体金免疫层析法[9]、凝胶渗透色谱法[10]和高效液相色谱-质谱联用法[11-13]。
随着人们生活水平的提高,人们在追求牛奶营养的同时,也开始对牛奶口味有了更多的需求,由此国内市场上孕育而生出各种口味的调制乳。2015年国内伊利和蒙牛两大乳品企业的财务报告显示其2014年销售额均突破500亿人民币,调制乳在其销售份额中占有重要比重。然而现阶段我国主要使用GB/T 22966-2008《牛奶和奶粉中16种磺胺类药物残留量的测定液相色谱-串联质谱法》[14]检测牛奶中磺胺类药物残留。该方法检测生乳、灭菌乳、巴氏杀菌乳等样本时能够获得较好的实验结果,但在检测基质复杂的调制乳样本时,杂质不能被有效地去除,提取效率低,影响了检测结果的准确性。此外,针对调制乳中磺胺类药物残留的检测方法报道也很少,所以研究调制乳中磺胺类药物残留检测方法非常有必要,也具有实际意义。乳品加工企业依据GB 19301-2010《食品安全国家标准生乳》规定对牛奶中磺胺药物残留进行检测,一般遇到磺胺类药物残留量超标,绝大多数是由磺胺嘧啶、磺胺甲基嘧啶或磺胺二甲基嘧啶3种磺胺类药物超标造成的,故本研究主要分析上述3种磺胺类药物。本研究成果可以作为对国家标准检测方法的补充,以保证调制乳中磺胺类药物处于有效监控中,为消费者的健康提供保障。
1 实验部分
1.1 主要仪器
UPLC-TQD液相色谱-串联质谱仪:配有电喷雾离子(ESI)源(美国 Waters公司);Milli-Q超纯水器(美国Millipore公司);漩涡混合仪(德国IKA公司);离心机(上海力申科学仪器有限公司);氮吹仪(美国Organomation公司);固相萃取装置(美国SUPELCO公司)。
1.2 试剂与耗材
甲酸、乙酸(色谱纯,天津市福晨化学试剂厂);甲醇、乙腈(HPLC纯,Fisher公司);磺胺嘧啶、磺胺甲基嘧啶和磺胺二甲基嘧啶标准品(纯度≥95%,DR.Ehrenstorfer公司);HLB固相萃取小柱(60 mg,3 mL,Waters公司);有机系滤头(0.22 μm,Waters公司);实验室用水为Milli-Q超纯水器制备的超纯水。
标准溶液:准确称取适量的磺胺标准品,用甲醇配制100 mg/L的标准储备液,-20℃冰箱中储存。使用时先用甲醇稀释成1 mg/L的中间液,再用阴性基质溶液稀释成系列标准工作液。
1%乙酸水溶液:将1 mL乙酸置于100 mL容量瓶中,用水定容至刻度,混匀后得到。0.1%甲酸水溶液:取0.1 mL甲酸,用水定容至100 mL,混匀后得到。20%甲醇水溶液:取20 mL甲醇,用水定容至100 mL,混匀后得到。
1.3 样品处理与测定
1.3.1 样品制备与保存
取均匀样品约250 g装入洁净容器作为试样,标明标记,密封至4℃下保存。
1.3.2 样品提取
称取2 g(精确到0.01 g)待测调制乳样品于50 mL离心管中,加入4 mL 1%乙酸水溶液,置于漩涡混合仪上高速漩涡2 min;再加入12 mL甲醇,高速漩涡2 min 后离心(6 000 r/min ,8 min),取离心后的上清液于鸡心瓶中备用。
1.3.3 样品净化
将提取液于50℃旋转蒸发至5 mL以下,用6 mL 1%乙酸水溶液分两次洗涤鸡心瓶,将洗液与处理好的样品混匀,加入到已活化的HLB固相萃取柱(HLB固相萃取柱使用前依次用4 mL甲醇、4 mL水过柱)中,控制流速低于2 mL/min,待滤液全部通过后,用3 mL水淋洗并抽干HLB固相萃取柱;最后用4 mL甲醇洗脱,收集洗脱液,得到净化液。将净化液于50℃水浴中用氮气吹干,再加入1 mL 20%甲醇水溶液,涡旋溶解后经0.22 μm有机系滤膜过滤,滤液供UPLC-MS/MS检测。
1.4 UPLC-MS/MS 条件
1.4.1 色谱条件
色谱柱:ACQUITY UPLC HSS T3柱(100 mm×2.1 mm,1.8 μm);流动相:乙腈(A)和0.1% 甲酸水溶液(B);流速:0.3 mL/min;梯度洗脱程序:0 ~1 min,25%A ~60%A(曲线6:线性梯度);1~3 min,60%A(曲线6:线性梯度);3~5 min,60%A ~25%A(曲线1:台阶梯度)。柱温:30 ℃;进样量:5 μL。
1.4.2 质谱条件
离子化模式为电喷雾正离子(ESI+)模式;质谱扫描方式为多反应监测(MRM)模式;电离电压为3.00 kV;离子源温度为120℃;去溶剂气温度为380℃;去溶剂气为氮气,流量为650 L/h;碰撞气为高纯氩气,流量为20 L/h。锥孔电压、碰撞能量及定性和定量离子等质谱参数如表1所示。
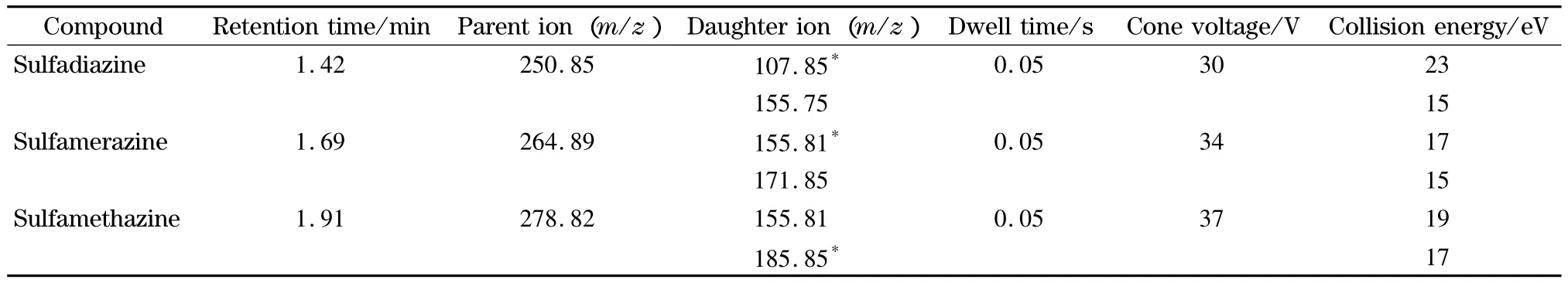
表1 3种磺胺类药物的保留时间和质谱参数Table 1 Retention times and mass spectrometric parameters of the three sulfonamides
2 结果与讨论
2.1 提取剂的选择
目前常用乙腈、乙酸乙酯、甲醇、二氯甲烷等有机溶剂或盐酸、三氯乙酸、高氯酸等酸溶液提取牛奶中残留的磺胺类药物。乙腈和甲醇均能很好地提取样品中的磺胺类药物,但乙腈沸点较甲醇高,提取浓缩耗时长,毒性相对较大,且乙腈提取磺胺甲恶唑的效率不如甲醇[6]。林海丹等[15]使用乙酸乙酯对样品进行提取时发现容易出现乳化现象,提取后样品杂质较多,存在干扰,不适合鲜奶样品的提取。本研究前期也曾使用乙酸乙酯对朱古力牛奶、咖啡牛奶和花生牛奶样品进行提取,检测后发现乙酸乙酯对样本中3种磺胺药物的提取效率偏低,磺胺嘧啶的回收率只有50%左右,磺胺甲基嘧啶和磺胺二甲基嘧啶的回收率在60%左右,说明乙酸乙酯也不适合调制乳样品的提取。
由于磺胺类药物呈弱碱性,增加酸性可以增大磺胺在水中的溶解性[16]。实验采用先加入乙酸溶液的提取方式将磺胺类药物从牛奶中解离出来,然后用甲醇进一步提取,在提取过程中去除样品中的蛋白质等杂质。实验使用1%乙酸水溶液和甲醇先后对样品进行提取,能够有效地提取出样品中的3种磺胺类药物。
2.2 固相萃取柱的选择
张志刚[17]曾使用0.1%乙酸乙腈溶液提取猪肉中的13种磺胺药物,然后浓缩上机供UPLC-MS/MS检测,取得了较好的实验结果。但调制乳样品基质复杂,样本种类繁多,单纯地仅仅是提取、浓缩,不进行固相萃取柱净化,很难获得满意的实验结果。实验考察了用乙腈和乙酸乙酯(4∶1,v/v)混合溶液提取样本,然后将样本经过除脂、浓缩、复溶后上机检测,结果显示样本的噪声较大,加标浓度在5 μg/kg以下时,样本的回收率和重复性均较差,说明使用固相萃取柱对调制乳样本进行净化非常必要。
目前用于磺胺类药物检测时使用的固相萃取柱主要有C18萃取柱、阳离子萃取小柱、氨基柱萃取柱、碱性氧化铝萃取柱和HLB萃取柱等。实验对比了使用C18萃取柱、HLB萃取柱和混合阳离子交换柱的净化效果,结果显示:使用混合阳离子萃取柱对样本进行净化,以5%氨化甲醇洗脱,最终3种磺胺类药物的回收率均低于10%,可能是1%乙酸溶液不能很好地使磺胺类药物富集在阳离子交换小柱上;使用C18萃取柱和HLB萃取柱净化,以甲醇洗脱,HLB萃取柱的净化效果比C18萃取柱好。另外,杨方等[18]的研究指出,不同批次的C18萃取柱在相同条件下检测磺胺类物质获得结果的重现性不够理想;吴银良等[19]采用C18柱和氨基柱组合提取牛奶中的磺胺类药物,虽然获得了较好的结果,但使用了2支萃取柱净化样品,实验步骤较为繁琐,也增加了实验费用。因此本文最后采用HLB萃取柱作为净化柱。
2.3 基质效应
液相色谱-电喷雾质谱联用方法中产生基质效应的影响机制目前尚不清楚,但通常认为共提物基质成分与待测目标物在色谱柱出口电离竞争是引起基质效应的主要机理[20]。其结果会降低或增加目标离子的生成效率及离子强度,从而影响测定结果的精密度和准确度。实验采用阴性样品基质溶液与流动相分别配制标准溶液,在相同条件下进行UPLC-MS/MS检测。结果显示:阴性样品基质对磺胺嘧啶和磺胺甲基嘧啶具有增强效应,对磺胺二甲基嘧啶具有减弱效应。为了消除基质效应的影响,实验采用阴性样品基质配制标准溶液来定量调制乳中的3种磺胺类药物,实验结果令人满意。
2.4 流动相的选择
因磺胺类药物结构中含有-NH2,在流动相中加入酸可以增加其离子化效率,同时pH值也对磺胺类化合物在色谱柱上的保留有重要影响。实验分别使用0.1%甲酸水溶液和0.1%乙酸水溶液作为流动相做对比实验。结果显示:加入两种酸时,3种磺胺药物均有很好的响应;但以0.1%乙酸水溶液作为流动相时,3种磺胺类药物的保留时间显著提前,色谱峰不能完全分离;而以0.1%甲酸水溶液作为流动相时,3种磺胺类药物能够完全分离。因此本实验选择以0.1%甲酸水溶液作为流动相。
2.5 标准曲线和线性范围
配制质量浓度分别为 1、2、5、10、20、50 和 100 μg/L的磺胺嘧啶、磺胺甲基嘧啶和磺胺二甲基嘧啶混合标准溶液,用超高效液相色谱-串联四极杆质谱仪对标准工作液中磺胺嘧啶、磺胺甲基嘧啶和磺胺二甲基嘧啶进行检测。以定量离子的峰面积Y对其质量浓度X(μg/L)进行线性回归,得到磺胺嘧啶、磺胺甲基嘧啶和磺胺二甲基嘧啶的标准曲线(见表2),其线性响应范围完全满足测定的要求,证明该方法定量测定的可靠性。

表2 3种磺胺类药物的线性方程和相关系数(R2)Table 2 Linear equations and correlation coefficients(R2)of the three sulfonamides
2.6 回收率和精密度
由于目前市场上调制乳的种类繁多,实验仅选取朱古力牛奶、咖啡牛奶和花生牛奶3种具有代表性的调制乳产品进行回收率和精密度的验证实验。
取不含磺胺嘧啶、磺胺甲基嘧啶和磺胺二甲基嘧啶的牛奶作为空白样品,向空白样品中添加1、2和10 μg/kg的磺胺嘧啶、磺胺甲基嘧啶和磺胺二甲基嘧啶标准品,得到加标样品,按照1.3节的条件进行样品处理,1.4节的条件进行测定,每个加标水平平行测定6个样品,3种磺胺类药物的加标回收率及相对标准偏差(RSD)见表3。基质标准溶液、样品空白和样品空白加标的磺胺嘧啶、磺胺甲基嘧啶和磺胺二甲基嘧啶色谱图见图1。
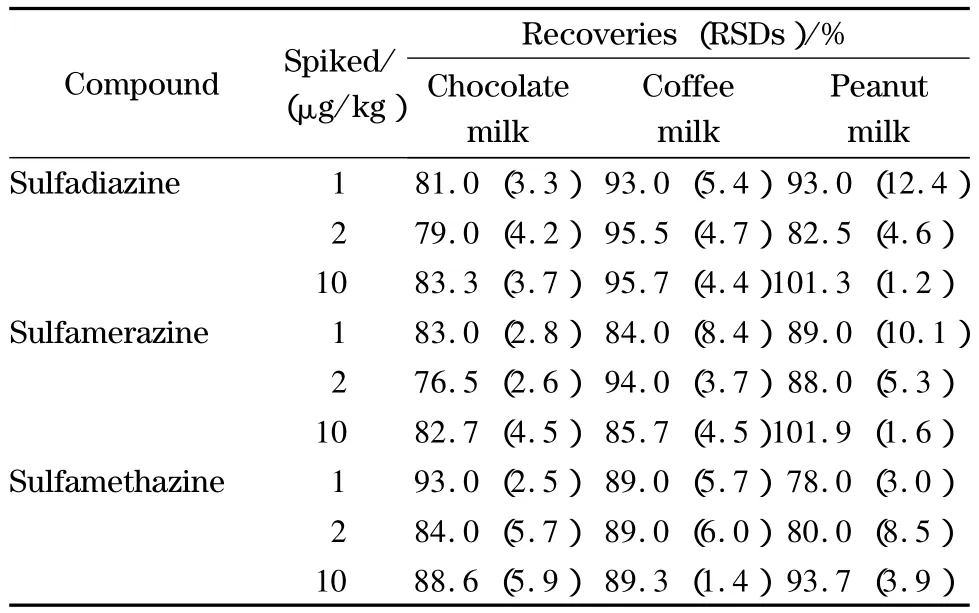
表3 空白样品中3种磺胺类药物的加标回收率及相对标准偏差(n=6)Table 3 Recoveries and RSDs of the three sulfonamides spiked in blank samples(n=6)
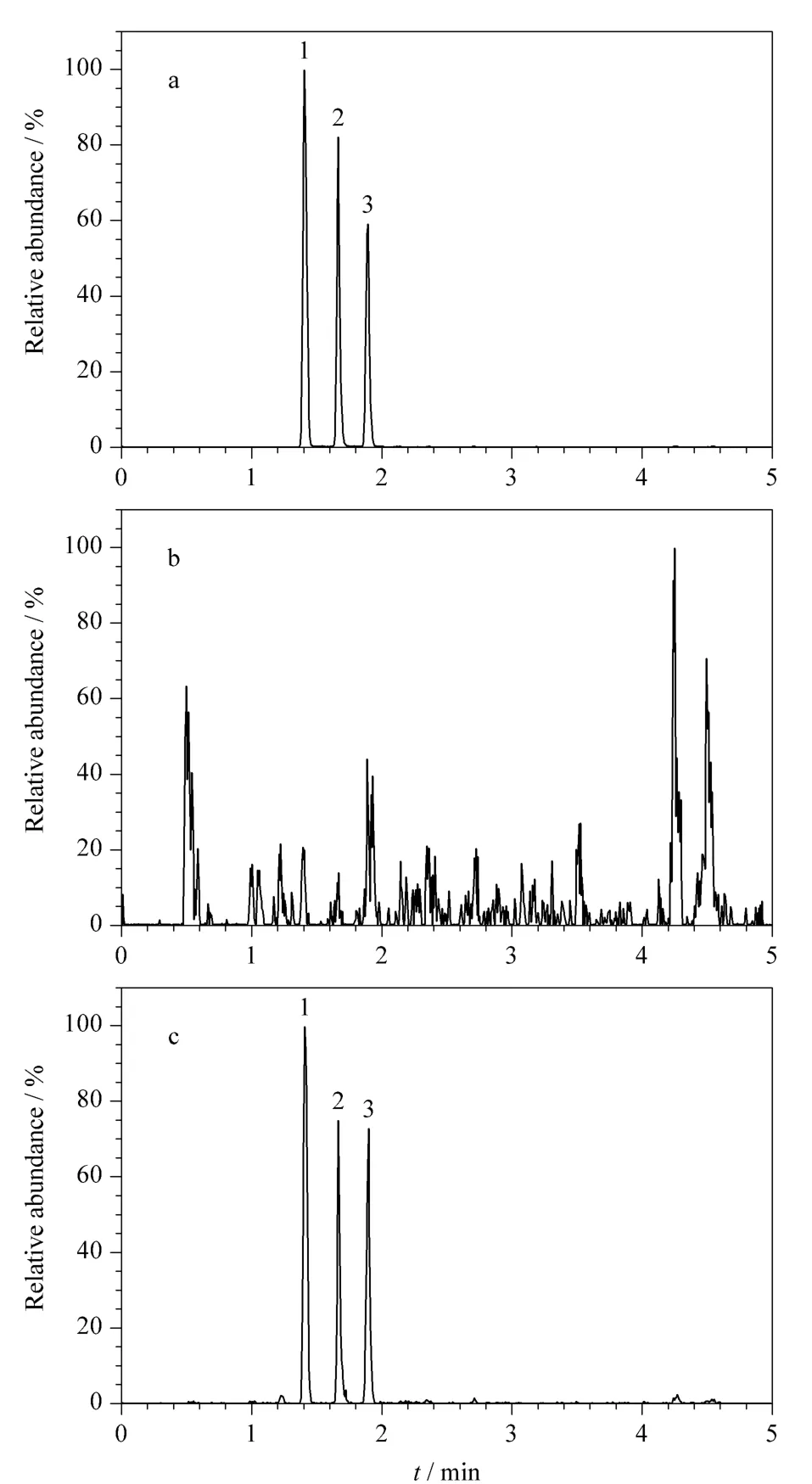
图 1 (a)阴性基质混合标准工作液(20 μg/L)、(b)空白调制乳样品和(c)加标调制乳样品(1 μg/kg)的色谱图Fig.1 Chromatograms of(a)a mixed standard working solution in negative matrix (20 μg/L),(b)a blank modified milk sample and (c)a spiked modified milk sample (1 μg/kg)
2.7 检出限
检出限(LOD)和定量限(LOQ)是任何方法论证的重要组成部分,国际纯粹与应用化学联合会(IUPAC)对检出限的定义为某特定方法在给定的置信度内可从样品中检出待测物质的最小浓度或量。LOD和LOQ受分离条件的影响非常显著,如色谱柱、试剂,尤其是仪器与数据处理系统。仪器的改变,尤其是泵系统和检测器,或使用被污染的试剂将使信噪比产生较大的变化(尤其影响基线噪声和漂移)。
首先用阴性基质配制标准工作液,然后按照UPLC-MS/MS条件对该工作液进行检测,算出3种磺胺类药物理论上的检出限(S/N=3时的浓度)和定量限(S/N=10时的浓度),结果见表4。检测过程中受前处理操作等因素的影响,往往方法的定量限与理论上的定量限有所差异,宋伟等[21]指出定量限为在获得满意的回收率和相对标准偏差条件下可以检测到的样品溶液中目标化合物的最低浓度。实际检测过程中添加3种磺胺类药物的浓度为1 μg/kg时,回收率和精密度均取得较为满意结果,因此将1 μg/kg作为方法的定量限。
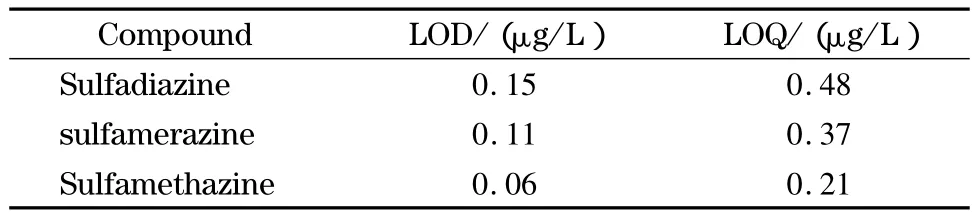
表4 UPLC-MS/MS测定调制乳中3种磺胺类药物的检出限和定量限Table 4 LODs and LOQs for the three sulfonamides in modified milk by UPLC-MS/MS
3 结论
调制乳经乙酸水溶液和甲醇提取、沉淀蛋白质,提取液经HLB固相萃取柱净化、甲醇洗脱,UPLCMS/MS测定,外标法定量。实验方法简便、快速、准确、实用,结果令人满意,适用于调制乳中磺胺嘧啶、磺胺甲基嘧啶和磺胺二甲基嘧啶的确证检测。
此外,GB/T 22966-2008中其他13种磺胺类药物也能有效地被1%乙酸水溶液和甲醇提取,本文建立的方法在固相萃取柱的选择、样品的富集和洗脱方面和GB/T 22966-2008方法类似,作为对国家标准方法的补充,也可以用于调制乳中其他13种磺胺类药物的检测。
[1] Xu G W,Kong X H,Li J H,et al.Acta Agriculturae Boreali-Occidentalis Sinica(徐国伟,孔祥虹,李建华,等.西北农业学报),2008,17(1):42
[2] Wan C H,Long Z X.Modern Scientific Instruments(万春花,龙洲雄.现代科学仪器),2008(2):99
[3] Wang W H,Han Z J,Wei X J,et al.Journal of Anhui Agricultural Sciences(王伟华,韩占江,魏新军,等.安徽农业科学),2006,34(6):1101
[4] Huang D M,Huang X Y,Gu R R,et al.Chinese Journal of Chromatography(黄冬梅,黄宣运,顾润润,等.色谱),2014,32(8):874
[5] Liu J H,Sun Z Z,Huang X L,et al.Chinese Journal of Chromatography(刘菁华,孙振中,黄雪玲,等.色谱),2015,33(4):434
[6] Li X D,Xian Q M,Liu H L,et al.Chinese Journal of Analytical Chemistry(李学德,鲜啓明,刘红玲,等.分析化学),2010,38(3):429
[7] Zhao H X,Liu H P,Yan Z Y.Chinese Journal of Chromatography(赵海香,刘海萍,闫早婴.色谱),2014,32(3):294
[8] Wu C Q,Lei J M,Li Y L,et al.Chinese Journal of Chromatography(吴翠琴,雷金妹,李韵灵,等.色谱),2014,32(12):1362
[9] Han J,Liu E M,Wang S,et al.Modern Food Science and Technology(韩静,刘恩梅,王帅,等.现代食品科技),2011,27(5):603
[10] Ouyang Y L,Xie W P,Huang Y Y,et al.Chinese Journal of Public Health(欧阳燕玲,谢维平,黄盈煜,等.中国公共卫生),2009,25(5):610
[11] Ma Q,Yu Q W,Luo Y B,et al.Chinese Journal of Chromatography(马乔,余琼卫,罗彦波,等.色谱),2011,29(7):624
[12] Yan C R,Zhang X Q,Lin H,et al.Meat Research (颜春荣,张晓强,林慧,等.肉类研究),2013,27(10):17
[13] Gong S S,Gu X,Cao H,et al.Journal of Instrumental Analysis(贡松松,顾欣,曹慧,等.分析测试学报),2014,33(12):1342
[14] GB /T 22966-2008
[15] Lin H D,Xie S X,Feng D X,et al.Journal of Instrumental Analysis(林海丹,谢守新,冯德雄,等.分析测试学报),2003,22(1):94
[16] Sun J W,Zhao X H.Food Science(孙晶玮,赵新淮.食品科学),2007,28(6):256
[17] Zhang Z G.Meat Research(张志刚.肉类研究),2013,27(2):13
[18] Yang F,Li Y P,Qian J,et al.Analysis and Testing Technology and Instruments(杨方,李耀平,钱疆,等.分析测试技术与仪器),2004,10(4):234
[19] Wu Y L,Zhao L,Liu Y J,et al.Chinese Journal of Chromatography(吴银良,赵莉,刘勇军,等.色谱),2007,25(5):728
[20] Wang L Q,He L M,Zeng Z L,et al.Journal of Chinese Mass Spectrometry Society(王立琦,贺利民,曾振林,等.质谱学报),2011,32(6):321
[21] Song W,Hu Y Y,Han F,et al.Chinese Journal of Chromatography(宋伟,胡艳云,韩芳,等.色谱),2013,31(12):1161
猜你喜欢
现代畜牧科技(2021年10期)2021-11-19 08:42:36
昆明医科大学学报(2021年8期)2021-08-13 08:59:04
中国油脂(2020年3期)2020-04-10 02:08:54
武警医学(2018年10期)2018-11-06 07:04:34
无机化学学报(2016年8期)2016-12-06 09:05:14
当代化工研究(2016年6期)2016-03-20 16:21:42
化学分析计量(2016年1期)2016-03-14 00:35:19
分析测试学报(2015年3期)2016-01-13 06:18:20
云南畜牧兽医(2015年4期)2015-02-28 21:26:11
无机化学学报(2014年3期)2014-02-28 17:30:55
