
烃类C—H键催化断裂活性受相邻基团的影响规律
2015-07-16 01:17秦玉才莫周胜王西阳张晓彤宋丽娟
石油化工高等学校学报 2015年6期
唐 亮, 李 强, 秦玉才, 万 海,,3, 莫周胜,王西阳, 张晓彤, 宋丽娟,
(1.辽宁石油化工大学辽宁省石油化工催化科学与技术重点实验室,辽宁抚顺113001;2.中国石油大学(华东)化学工程学院,山东青岛266555;3.中国石油抚顺石化公司石油三厂,辽宁抚顺113001)
稠油注空气低温氧化技术是一种新型高效的稠油三次开采技术,该技术在油藏条件下消耗掉空气中氧气,起到开采安全和增加地层能的作用[1-2]。稠油中的主要成分是含有惰性C—H键的烃类化合物,而目前大部分的惰性C—H键活化是在高温、高压以及强酸性条件下进行的,反应条件苛刻,并且活化效率不高。因此对烃类C—H键的活化深入研究对稠油注空气低温氧化机理的探究具有重要的理论指导意义。
目前,过渡金属催化C—H 键活化的方法具有反应条件温和、活化效率高等优点,使其得到广泛的应用[3-7],成为研究热点。对于过渡金属催化C—H键活化已有大量的研究报道。R.A.Periana等[8-9]分别使用Pt以及Pd催化剂在低温下实现了从甲烷到甲醇以及乙酸的高效转化。A.E.Shilov等[10]对Pt(II)催化C—H活化进行了研究,向含有甲烷的水溶液中加入Pt(IV),甲烷将选择性氧化生成甲醇和一氯甲烷。S.Murai[11]等以钌络合物作为催化剂,通过羰基的定位作用实现了芳香化合物邻位C—H键的活化。然而,到目前为止,在过渡金属催化剂的作用下,稠油中的烃类C—H键催化活化受其相邻基团影响规律的研究还较少。
在过渡金属催化烃类C—H键活化过程中,C—H键的断裂是最为困难的一步,因此,C—H键活化过程受C—H键断裂的影响最大。本文选取带有苯基的甲苯、乙苯、异丙苯,带有环己基的甲基环己烷、甲基环己烷、乙基环己烷、异丙基环己烷,带有正戊基的正己烷、正庚烷、2-甲基辛烷作为典型的芳香烃、环烷烃、饱和烃代表,并以醋酸铜作为模型催化剂,采用密度泛函理论研究了烃类中甲基、亚甲基、次甲基C—H键在醋酸铜催化作用下断裂活性受其相邻基团苯基、环己基、正己基的影响规律,为探究稠油中烃类C—H键活化规律提供理论依据。
1 计算方法
所有量化计算均在美国Accelrys公司的Materials Studio5.5软件包中的Dmol3模块下进行,采用基于广义梯度近似(GGA)的密度泛函理论,选取PW91交换相关泛函和DNP基组对反应物、中间体、过渡态和产物的几何构型进行优化,并通过振动频率分析确认稳定点和过渡态结构,进行IRC计算确认过渡态连着正确的反应物和产物。计算对电子未限制自旋,选取DSPP方法描述原子内层电子,计算精度为Fine。自洽迭代过程收敛参数设置为:总能量收敛至1.0×10-5Ha、自洽场收敛至1.0×10-6Ha、原子间作用力收敛至2.0×10-2Ha/nm、最大位移为5.0×10-4nm,为了加速收敛过程,Thermal Smearing值设为5.0×10-3Ha。
2 结果与讨论
2.1 烃类C—H键断裂过程的性质分析
图1为烃类C—H键的断裂位置示意图。采用分子模拟方法主要对图1中9种烃类的C—H键的键能进行计算。

图1 烃类C—H键断裂的过程示意图Fig.1 Scheme of cleavage process of C—H bond in hydrocarbons
表1给出了以上9种烃类的键能数据,可以看到甲苯、乙苯、异丙苯,甲基环己烷、乙基环己烷、异丙基环己烷,正己烷、正辛烷、2-甲基戊烷符合C—H键断裂规律,即甲基、亚甲基、次甲基上C—H键能逐渐降低,反映出其断裂活性依次增加,并且甲苯、乙苯、异丙苯的C—H键能分别为418.6、398.7、385.1kJ/mol,与 文 献 值[12-14](375.4~385.6、377.8、362.8kJ/mol)较为接近。这说明选择的模拟方法和参数设置是合理的。
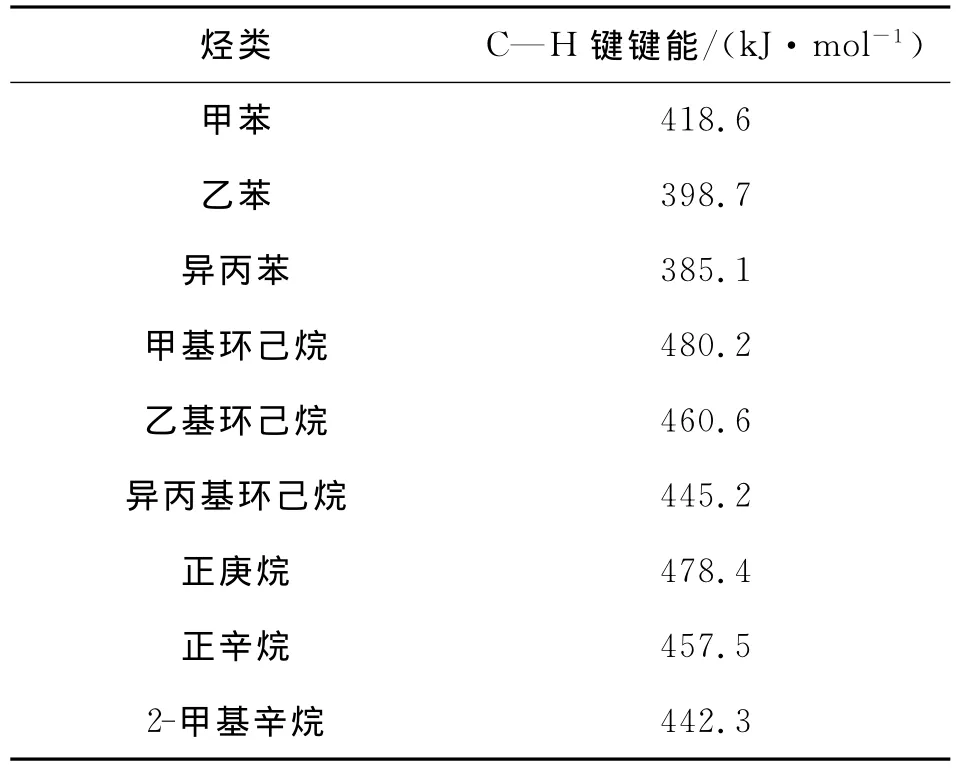
表1 各种烃类的C—H键键能Table1 The C—H bond energy of various kinds of hydrocarbon
2.2 不同烃类中的甲基C—H键催化断裂过程
图2为甲苯、甲基环己烷、正庚烷中甲基C—H键催化断裂过程的反应势能图。如图2所示,甲苯发生C—H键催化断裂过程,首先与醋酸铜发生吸附作用,形成甲苯络合物IM11,这一过程释放80.3 kJ/mol的热量。接下来,IM11发生氢转移反应,甲苯中的甲基C—H键逐渐断裂,O—H键逐渐形成。当C—H拉长到0.137nm和O—H缩短到0.129 nm时,反应到达反应能垒为80.0kJ/mol的过渡态TS11。之后,C—H键完全断裂,O—H键形成,得到中间体IM12,完成甲苯的C—H键断裂过程。而R21甲基环己烷和R31正庚烷中甲基发生C—H键断裂过程,则会与醋酸铜发生吸附作用,形成Cu…H键,得到中间体IM21和IM31,此时IM21和IM31的能量分别低于反应物85.2kJ/mol和109.9kJ/mol。随后,IM21和IM31的甲基上的H原子经历C—H断裂、Cu—H键形成的过渡态TS21和TS31,得到中间体IM22和IM32,C—H键完全断裂。IM21和IM31完成C—H键断裂过程的反应能垒分别为160.8、156.8kJ/mol。上述结果表明,与不同基团相连的甲基C—H键的催化断裂活性顺序:苯基>正己基>环己基。
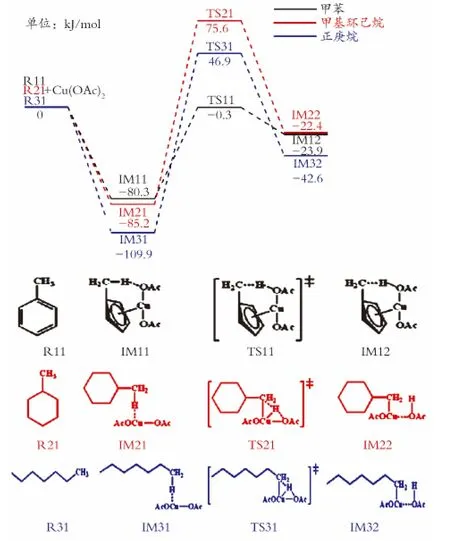
图2 甲苯、甲基环己烷、正庚烷中甲基C—H键催化断裂过程的反应势能图Fig.2 Energy profile of the catalytic cleavage of C—H bond of methyl in toluene,methylcyclohexane,n-heptane
图3为乙苯、乙基环己烷、正庚烷中亚甲基C—H键催化断裂过程的反应势能图。如图3所示,乙苯中的亚甲基与甲苯中的甲基C—H键催化断裂过程类似,乙苯首先与醋酸铜发生吸附作用,放出84.6kJ/mol的能量,得到络合物IM41。之后,IM41经历C—H键逐渐断裂和O—H键逐渐形成过程,越过能垒为72.9kJ/mol的过渡态TS41,形成C—H键断裂后的中间体IM42。乙基环己烷和正辛烷中的亚甲基C—H键发生催化断裂,首先与醋酸铜经历吸附过程,能量分别降低了94.9、83.0kJ/mol,得到反应中间体IM51和IM61。接下来,IM51和IM61发生C—H键断裂和Cu—H键形成过程,得到过渡态TS51和TS61。最后,TS51和TS61中的O—H键完全形成得到中间体IM52和IM62。乙基环己烷和正庚烷完成C—H键催化断裂过程的能垒分别为175.9、179.7kJ/mol。这说明,与不同基团相连的亚甲基C—H键的催化断裂活性顺序:苯基>环己基>正己基。
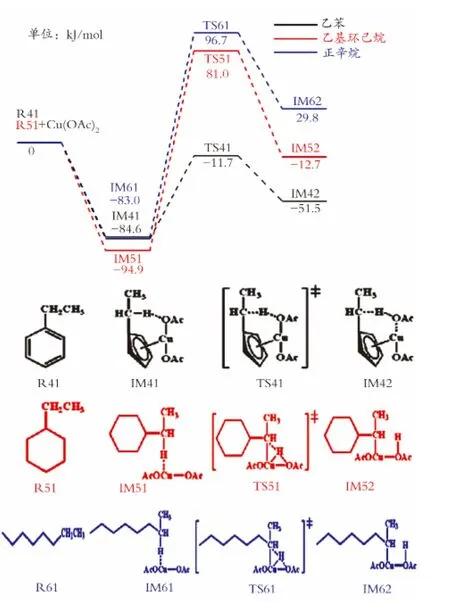
图3 乙苯、乙基环己烷、正辛烷中亚甲基C—H键催化断裂过程的反应势能图Fig.3 Energy profile of the catalytic cleavage of C—H bond of methylene in ethylbenzene,ethylcyclohexane,n-octane
图4为异丙苯、异丙基环己烷、2-甲基辛烷中次甲基上C—H键催化断裂过程的反应势能图。如图4所示,与上述几种烃类的C—H键催化断裂过程类似,异丙苯与醋酸铜发生吸附作用时放出86.2 kJ/mol的热量,形成络合物IM71,之后经历能垒为73.2kJ/mol的过渡态 TS71,然后形成中间体IM72,完成C—H键的断裂过程。异丙基环己烷和2-甲基辛烷与醋酸铜发生吸附作用,各放出103.5 kJ/mol和49.6kJ/mol的能量,之后分别越过能垒为234.0、160.9kJ/mol的过渡态 TS81和 TS91,得到C—H键断裂后的中间体IM82和IM92。通过比较这3种物质C—H键活化能垒可知,与不同基团相连的次甲基C—H键的催化断裂活性顺序:苯基>正己基>环己基。
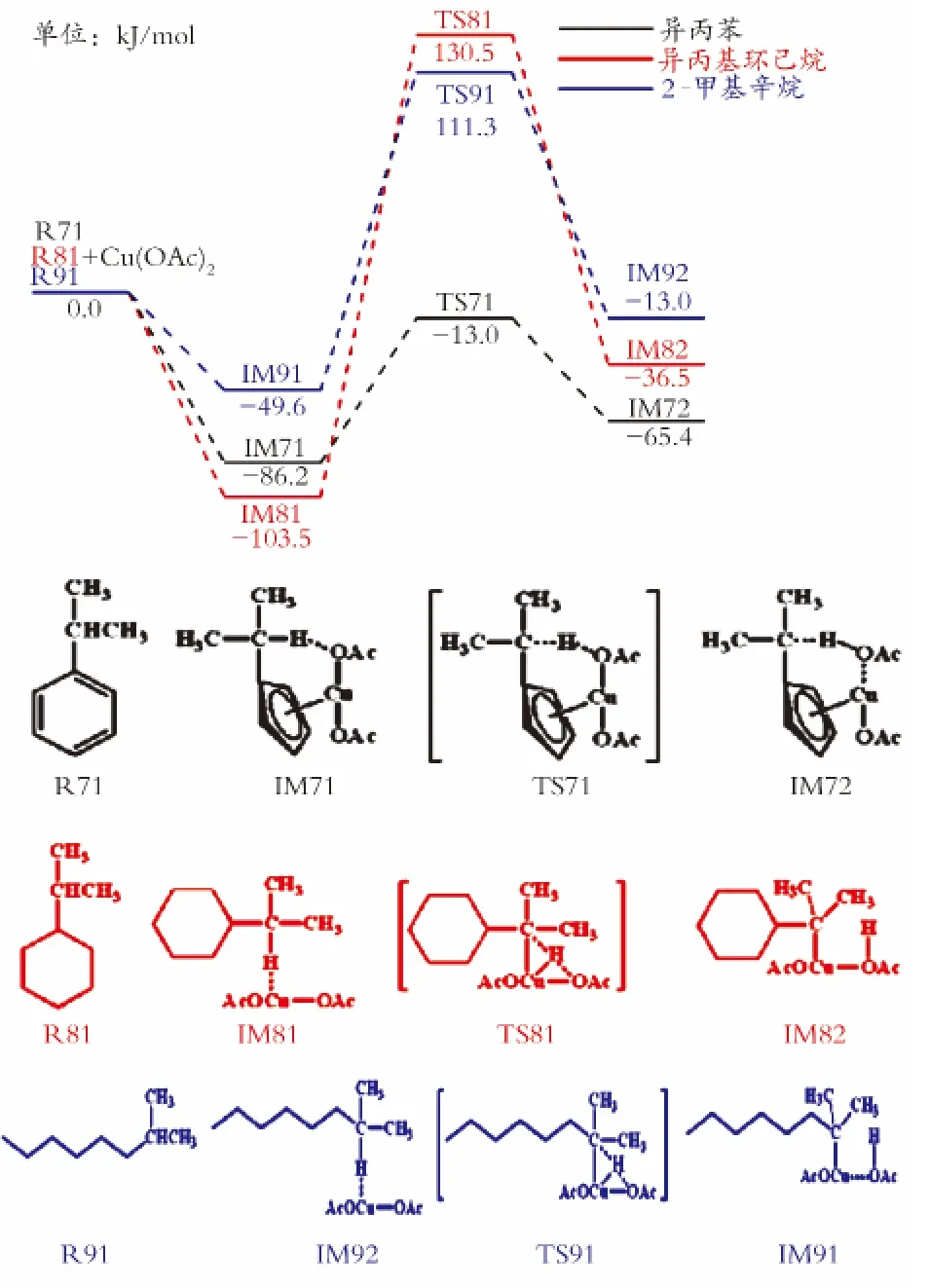
图4 异丙苯、异丙基环己烷、2-甲基辛烷中次甲基C—H键催化断裂过程的反应势能图Fig.4 Energy profile of the catalytic cleavage of C—H bond of methyne in isopropylbenzene,isopropylcyclohexane,2-methyloctane
2.3 不同烃类中的甲基C—H键催化断裂活性的理论解释
图5为吸附前后与C—H键相连的苯基、环己基以及正己基的电荷变化量(ΔQ)与C—H键催化断裂反应能垒的关系曲线。如图5所示,在烃类与催化剂发生吸附前后,与甲基、次甲基C—H键相连的苯基、环己基以及正己基的电荷变化量和C—H键催化断裂反应能垒呈线性关系,随着苯基、环己基以及正己基的电荷变化量增加,反应能垒逐渐降低,这是因为与C—H键相连的基团电荷量增加,使C—H键的离子性增加,C—H键断裂活性更高。因此对于甲基C—H键催化断裂活性顺序为:IM11>IM31>IM21,对于亚甲基C—H键催化断裂活性顺序为:IM41>IM51>IM61。
图6为C—H键上甲基数目与其催化断裂反应能垒的关系曲线。如图6所示,当C—H键与环己基相连时,随着C—H键上甲基数目的增加,C—H键催化断裂反应能垒逐渐增加,这是因为C—H键上甲基数目增多,会导致C—H键催化断裂反应空间位阻增加,从而使C—H键断裂的活性降低。因此,对于与环己基相连的C—H键催化断裂活性顺序为:IM21>IM51>IM81。
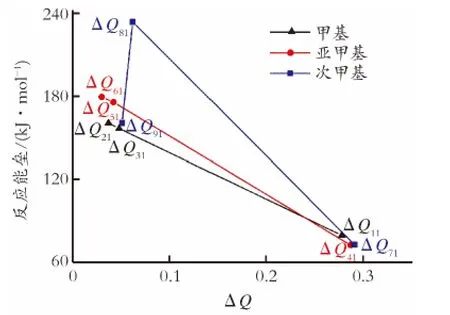
图5 吸附前后与C—H键相连的苯基、环己基以及正己基的电荷变化量与C—H键催化断裂反应能垒的关系曲线Fig.5 The relationship curve between the changes of charge value of phenyl,cyclohexyl and n-hexyl which are linked with C—H bond during adsorption process and the energy barrier of C—H bond catalytic cleavage
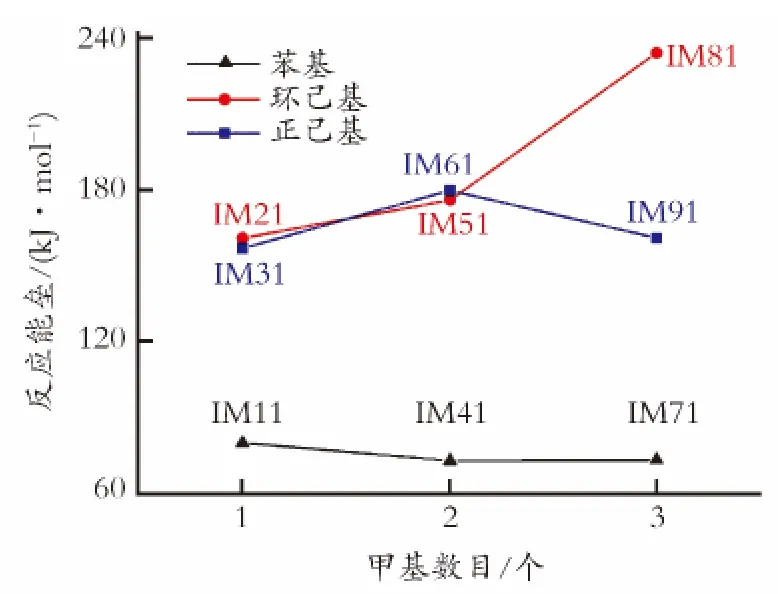
图6 C—H键上甲基数目与其催化断裂反应能垒的关系曲线Fig.6 The relationship curve between the number of methyl which linked C—H bond and the the energy barrier of C—H bond catalytic cleavage
从图6可以看到,对于次甲基C—H键催化断裂活性顺序为:IM71>IM91>IM81。与苯基、环己基、正己基的电荷变化量顺序不一致,这可能是因为环己基与次甲基C—H键相连时的空间位阻高于正己基,因此C—H键断裂最困难。由图6还可以看出,对于与苯基相连的C—H键催化断裂活性顺序为:IM41>IM71>IM11,与C—H键上甲基数目变化的趋势不一致。这是因为苯基与甲基C—H键相连时,电荷变化量低于与亚甲基、次甲基相连时的电荷变化量,因此,甲基C—H键较亚甲基、次甲基难断裂。而对于与正己基相连的C—H键催化断裂活性顺序为IM31>IM91>IM61,也与C—H键上甲基数目变化的趋势不一致,这是因为正己基与次甲基C—H键相连时,电荷变化量高于与亚甲基相连时的电荷变化量,因此,次甲基C—H键较亚甲基容易断裂。
3 结论
通过对烃类C—H键在醋酸铜催化作用下断裂活性受其相邻基团影响规律的研究,结果表明,与不同基团相连的甲基和次甲基C—H键的催化断裂活性顺序:苯基>正己基>环己基;而亚甲基C—H键的催化断裂活性顺序:苯基>环己基>正己基。
通过对烃类C—H键催化断裂活性的理论解释,结果表明,烃类与催化剂发生吸附前后,与C—H键相连的基团电荷变化量越大,C—H键的离子性越强,C—H键断裂活性越高。另外,C—H键上甲基数目越多,C—H键催化断裂反应空间位阻越大,C—H键断裂的活性越低。
[1]崔盈贤,唐晓东,李晶晶,等 稠油氧化自生表面活性剂乳化降黏实验研究[J].西南石油大学学报:自然科学版,2013,35(3):154-159.Cui Yingxian,Tang Xiaodong,Li Jingjing,et al.Experimental study on reducing viscosity by emulsification of surfactants from heavy oil oxidation[J].Journal of Southwest Petroleum University(Science & Technology Edition),2013,35(3):154-159.
[2]王焕梅,唐晓东,孟科全,等.稠油注空气催化氧化采油催化剂的制备与评价[J].精细化工,2009,26(6):566-569.Wang Huanmei,Tang Xiaodong,Meng Kequan,et al.Preparation and evaluation of catalyst for heavy oil injected air catalytic oxidation production[J].Fine Chemicals,2009,26(6):566-569.
[3]Jones W D.Isotope effects in C—H bond activation reactions by transition metals[J].Accounts Chem.Res.,2003,36(2):140-146.
[4]Arndtsen B A,Bergman R G,Mobley T A,et al.Selective intermolecular carbon-hydrogen bond activation by synthetic metal complexes in homogeneous solution[J].Accounts Chem.Res.,1995,28(3):154-162.
[5]Tellers D M,Yung C M,Arndtsen B A,et al.Electronic and medium effects on the rate of arene ch bond activation by cationic Ir(III)complexes[J].J.Am.Chem.Soc.,2002,124(7):1400-1410.
[6]Niu S,Hall M B.Inter-and intramolecular C—H activation by a cationic iridium(III)center via oxidative-addition reductive-elimination andσ-bond metathesis pathways[J].J.Am.Chem.Soc.,1998,120(24):6169-6170.
[7]Fu R,Bercaw J E,Labinger J A.Intra-and intermolecular C—H activation by bis(phenolate)pyridineiridium(III)complexes[J].Organometallics,2011,30(24):6751-6765.
[8]Periana R A,Taube D J,Gamble S,et al.Platinum catalysts for the high-yield oxidation of methane to a methanol derivative[J].Science,1998,280(5363):560-564.
[9]Periana R A,Mironov O,Taube D,et al.Catalytic,oxidative condensation of CH4to CH3COOH in one step via CH activation[J].Science,2003,301(5634):814-818.
[10]Shilov A E,Shul'pin G B.Activation of C—H bonds by metal complexes[J].Chem.Rev.,1997,97(8):2879-2932.
[11]Murai S,Kakiuchi F,Sekine S,et al.Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins[J].Nature,1993,366(9):529-531.
[12]Brocks J J,Beckhaus H D,Beckwith A L,et al.Estimation of bond dissociation energies and radical stabilization energies by ESR spectroscopy[J].The Journal of Organic Chemistry,1998,63(6):1935-1943.
[13]Tumanov V,Kromkin E,Denisov E.Estimation of dissociation energies of C—H bonds in oxygen-containing compounds from kinetic data for radical abstraction reactions[J].Russ.Chem.B,2002,51(9):1641-1650.
[14]Muralha V S,Borges Dos Santos R M,Martinho Simões J A.Energetics of alkylbenzyl radicals:a time-resolved photoacoustic calorimetry study[J].The Journal of Physical Chemistry A,2004,108(6):936-942.
猜你喜欢
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
化工管理(2021年7期)2021-05-13
环境保护与循环经济(2020年4期)2020-06-08
化工进展(2020年1期)2020-01-15
中国特种设备安全(2019年1期)2019-03-13
安徽化工(2019年1期)2019-03-04
环境科技(2015年2期)2015-11-08
郑州大学学报(工学版)(2014年4期)2014-03-25

- 石油化工高等学校学报的其它文章
- 新型柴油降凝剂的合成方法
- 降低CO2驱混相压力的发展现状
- 《石油化工高等学校学报》征稿启事