
犬瘟热病毒GZ-Z株H基因原核表达载体的构建
2015-07-12 17:43:14薛磊
贵州畜牧兽医 2015年6期
薛 磊
(贵州省遵义市湄潭县农牧局,贵州湄潭564100)
犬瘟热病毒GZ-Z株H基因原核表达载体的构建
薛 磊
(贵州省遵义市湄潭县农牧局,贵州湄潭564100)
利用实验室构建的含有犬瘟热病毒H基因的pMD18-H质粒,根据其序列设计带有BamHⅠ 和HindⅢ 酶切位点的引物,对H基因ORF进行PCR扩增,得到约1 949 bp的片段。将该片段克隆到pMD18-T载体内,用BamHⅠ 和HindⅢ 进行双酶切鉴定、质粒PCR鉴定,筛选出阳性克隆。将阳性克隆再用BamHⅠ 和HindⅢ 进行双酶切,纯化回收CDV H基因ORF片段;将原核表达载体pET32a(+)用同样的方法酶切,回收载体片段,并用T4连接酶将以上两回收片段连接,构建原核表达载体质粒pET32-H,后经BamHⅠ 和HindⅢ 双酶切、质粒PCR、测序进行系列鉴定。结果表明,pET32-H原核表达质粒构建成功,其中插入片段大小为1 842 bp,可编码607个氨基酸残基。该实验为下一步H蛋白的原核表达及单克隆抗体的制备奠定了基础。
犬瘟热病毒;原核表达;PCR;H基因
犬瘟热(Canine Distemper,CD)是由犬瘟热病毒(Canine Distemper Virus,CDV)引起的一种高度接触性、致死性、急性病毒传染病[1, 2],是目前威胁养犬业、毛皮动物饲养和野生动物保护的主要疫病之一[3]。本病广泛分布于世界各地,而且容易继发其他细菌、病毒的混合感染和二次感染,死亡率可高达30%~80%[4]。犬瘟热的临床特征为早期表现双相热、急性鼻卡他,随后以支气管炎、卡他性肺炎、严重的胃肠炎、结膜炎、角膜炎[5]和神经症状为特征。少数病例出现鼻部和脚垫的高度角质化。犬瘟热的临床症状受毒株毒力、环境、宿主年龄、宿主免疫状况和感染种类的影响。在所有易感宿主中,呼吸系统、胃肠道、中枢神经系统和体表组织是最易感染的部位。
CDV在分类上属副粘病毒科,麻疹病毒属[6]。它与麻疹病毒(MV)、牛瘟病毒(RPV)、小反刍兽疫病毒(PPRV)、海豹瘟热病毒(PDV)、海豚瘟热病毒(PMV)同属于副粘病毒科(Paramyxoviridae)麻疹病毒属(Morbillivirus) 的成员[7,8]。CDV为不分节段的负链RNA病毒,呈圆形或不正形,有时呈长丝状,直径100~300 nm不等,大小差异较大。CDV的核衣壳由基因组与蛋白N组成,呈螺旋状盘曲在病毒粒子的中央,外面裹着1层脂质的囊膜,囊膜上密布有纤突[9]。多呈球形或不规则形,亦有畸形和长丝状的病毒粒子,粒子中心含有宽径为15~17 nm的螺旋形核衣壳,由基因组RNA和许多核衣壳蛋白亚单位组成。核衣壳外面包被1层由细胞膜和脂质衍生而来的双层脂质囊膜。膜上排列有由H和F 2种糖蛋白组成的杆状纤突,该纤突只含血凝素,无神经氨酸酶[7]。
CDV的基因组包含15 690个核苷酸,由7个基因组成,从3’端到5’端依次为:3’端前导序列、核衣壳蛋白基因N、磷蛋白基因P、膜蛋白基因M、融合蛋白基因F、血凝素蛋白基因H、大蛋白基因L和5’端前导序列。各基因上有自己的开放性阅读框架,其中P基因包含2个部分重叠的ORF。其中H蛋白基因由1 944~1 946个核苷酸组成,它的mRNA的5’端有20个核苷酸的非编码区, 3’端有100个或更多核苷酸的非编码区,在5’端或3’端的非编码区序列中没有发现稳定的茎环结构。CDV H蛋白有广泛的糖基化位点,糖基化位点末被蛋白酶剪切。在麻疹病毒属所有结构蛋白中,血凝蛋白(H蛋白)作为宿主免疫系统的目标抗原,其基因序列的变异程度是最高的[10],在免疫压力作用下,CDV抗原表位可能发生漂移,尤其是较大的H蛋白极易发生变异,从而引起CDV毒力变化[11]。H蛋白是病毒侵袭宿主所必需,同时H蛋白上存在许多中和性抗原表位,是诱导机体产生中和抗体的主要抗原之一,在抗CDV免疫中起着重要作用[12]。H蛋白决定CDV宿主的特异性,并协助F蛋白使CDV以囊膜与宿主细胞膜发生融合的方式进入宿主细胞。
CD呈世界性分布,并且能在种间进行传播流行,传播途径多元化,实验证明,传播的途径主要是呼吸道,其次是消化道,如通过飞沫、食物或不洁的医疗卫生用具以及眼结膜、口腔、鼻腔黏膜感染,另外还可以经阴道、直肠黏膜感染。CD康复犬可获得终身免疫力[13]。目前控制该病主要依赖于接种弱毒疫苗,并取得了显著的成效,然而随着使用的广泛,发现其存在许多缺陷和缺乏有效的检测疫苗免疫效果的方法[14,15]。没有吃初乳或母犬有很少或没有CDV抗体的少于6周龄的幼犬,对CDV易感,但3周龄之前接种的效果又不好,此时犬的抗体反应和细胞介导的反应还没有完全彻底地起作用,不能对CDV产生免疫应答,直到6周龄免疫系统才健全。
犬瘟热还没有可以治愈的药物,目前靠疫苗尤其是弱毒疫苗来控制本病,但疫苗存在一定的不安全性,同时犬瘟热病毒自身的基因发生突变,导致犬瘟热在各地时有发生。鉴于此,本实验根据实验室已构建并测序的含H基因的质粒序列,设计1对特异性引物,拟扩增CDV H ORF,并构建原核表达载体,为该病及其病原的进一步研究奠定基础。
1 材料
质粒:pMD18-H克隆质粒由贵州大学动物科学学院实验室鉴定保存;克隆载体:pMD18-T Vector(TA连接试剂盒),购自大连宝生物公司;表达载体:pET32a(+)由本实验室保存;菌株:E.coLiDH5α由贵州大学动物科学学院实验室保存;BL21(DE3) Competent Cell购自TIANGEN;TaKaRa La PCRTM Kit Ver.2.1(PCR扩增试剂盒),购自大连宝生物工程有限公司。
2 方法
2.1 H基因的扩增
2.1.1 引物的设计与合成:根据实验室已构建并测序的含H基因的质粒序列,利用Primer Premier 6.0 软件设计1对特异性引物:
CDV-H-ExS: 5’-CgCggATgCTCTCCTACCAAgACAAg-3’
CDV-H-ExAs: 5’-CCCAAgCTTTCAgggATTTgAACggTTACAT-3’
本实验预期扩增的基因片段大小为1 949 bp,引物由大连宝生物公司合成。
2.1.2 目的基因的扩增:以实验室鉴定保存的pMD18-H克隆质粒DNA为模板,用上述设计合成的特异性引物进行H基因的ORF扩增,反应体系如下:
pMD18-H质粒DNA 0.5 μL
CDV-H-ExAs 2.0 μL
CDV-H-ExS 2.0 μL
2×TaqPCR Mastermixture 12.5 μL
ddH2O 8.0 μL
将此反应体系按如下反应条件进行反应: 94 ℃预变性5 min; 94 ℃变性30 s,48 ℃ 复性1 min, 72 ℃延伸2 min,进行35个循环;最后72 ℃延伸 10 min。
反应结束后取出5 μL扩增产物进行1%琼脂糖凝胶电泳, 80 V电泳1 h后,于凝胶成像系统观察电泳结果。
2.2 H基因的克隆及鉴定
2.2.1 琼脂糖凝胶电泳:(1)称取琼脂糖1 g于200 mL 三角烧瓶中,加入1×TAE溶液100 mL。(2)在微波炉中加热溶化,完全溶解琼脂糖,直至无颗粒状结晶为止。(3)冷却至60 ℃左右,加入溴化乙锭10 μL,缓慢倒入胶槽中,尽量不要产生气泡。(4)室温静置30 min,待胶变硬、呈现乳白色且不透明时,除去梳子,取出胶板。(5)将配制好的 5×TAE 缓冲液稀释后倒入电泳槽。(6)取DL 2 000(Takara)marker 3 μL点样。(7)取PCR产物5 μL点在胶孔中。(8)将点好样的胶板放入电泳槽中,加样部位于负极,盖上槽板。(9)选择80 V电压进行电泳1 h。(10)取出于凝胶成像系统成像观察电泳结果,保存照片以备分析结果。
2.2.2 PCR产物的回收与纯化:参照E.Z.N.A.TMGel Extraction Kit(50)试剂盒进行胶回收,具体操作如下:(1)在紫外灯下切出含有目的DNA的琼脂糖凝胶,用纸巾吸尽凝胶表面的液体。此时应注意尽量切除不含目的DNA部分的凝胶,尽量减小凝胶体积,提高DNA回收率。(2)称量胶块重量,计算胶块体积。计算胶块体积时,以1 mg=1 μL进行计算。(3)加入与回收胶等体积的Binding Buffer(×P2),与60 ℃水中溶化。(4)将HiBind DNA column 放入2 mL Collection tube中。(5)将(1)中的溶液全部转入上述HiBind DNA column中,室温10 000 r/min 离心1 min。(6)倒掉液体,加入300 μL Binding Buffer(×P2),室温10 000 r/min离心 1 min。(7)倒掉液体,加入SPW 700 μL,室温10 000 r/min 离心1 min。(8)重复步骤(5)1次,倒掉液体,以大于13 000 r/min室温空离心2 min。(9)将HiBind DNA column放入新的1.5 mL Ep管中,在HiBind DNA column中央小心加入Elution Buffer 30 μL,室温静置10 min,以大于13 000 r/min室温空离心2 min。(10)将Ep管中的液体于4 ℃保存备用。
2.2.3 PCR产物与pMD18-T SimpIe vector的连接:将回收纯化的PCR产物与pMD18-T Simple vector连接构建重组质粒,在新的Ep管中依次加入下列成分:
回收的CDV-H 4.5 μL
pMD18-T Vector 0.5 μL
Ligation Solution Ⅰ 5.0 μL
将此反应体系16 ℃连接过夜,取出直接转化到E.coLiDH5α感受态细胞中。
2.2.4 大肠杆菌感受态细胞E.coLiDH5α的制备:(1)从37 ℃培养12~16 h的大肠杆菌E.coLiDH5α平板上无菌挑取生长良好的划线保存的单个菌落接种到10 mL LB肉汤中, 37 ℃, 200 r/min振荡培养过夜。(2)次日转移2 mL活化后的培养液至装有50 mL LB 肉汤的100 mL三角瓶中,继续 37 ℃振荡培养2~3 h,使其D600 nm达到0.4左右时将菌液取出立即冰水浴冷却10 min。(3)在无菌条件下将此细菌培养物转至灭菌过的50 mL离心管中。(4) 4 ℃ 3 500 r/min离心10 min以收集细胞,倾弃上清液并尽可能吸去所有培养液。(5)加入 25 mL 预冷的新配制的0.1%CaCl2溶液重悬菌体。(6)冰水浴30 min后, 4 ℃ 3 500 r/min离心10 min,弃上清并尽可能吸去所有培养液。(7)再用0.5~1.0 mL预冷的0.1%CaCl2溶液重悬菌体。(8)置 4 ℃冰箱12~24 h,即可应用于转化。新鲜的大肠杆菌感受态细胞可于 4 ℃储存24~48 h,但在储存的最初12~24 h转化效率最高。如1次制备的感受态不能用完,可冻存保留。其方法为:将感受态分装成 100 μL 1份,每份加20%体积的甘油保存液,置-70 ℃冰箱,低温保存可达3个月。
2.2.5 连接产物转化E.coLiDH5α感受态细胞[16]:(1)无菌状态下取新鲜感受态细胞100 mL,如果使用冻存的感受态细胞时,则将其从-70 ℃冰箱取出,握于手中,待其解冻后立即吸取100 mL转移至1.5 mL灭菌Ep管内。(2)待转化的质粒 1 μL (连接产物10 μL),加入感受态细胞中,轻轻旋转以混合内容物,在冰上放置30 min。(3)将离心管放到42 ℃水浴中热休克90 s,不要摇动离心管。(4)迅速将离心管转移到冰浴中,使细胞冷却 1~2 min。(5)加入800 μL无抗生素的LB培养基, 37 ℃温和振荡(转速200 r/min)培养45 min。(6)用无菌弯头玻璃铺菌器将200 μL菌液涂布于含有氨苄青霉素(终浓100 μg/L)的LB琼脂培养基平板上, 37 ℃倒置培养16 h。
2.2.6 质粒的小量制备(碱裂解法):本实验用灭菌Tip头分别挑取培养所见的白色单个菌落6个,记为1#~6#,分别接种于20 mL含AMP 10 μL的LB肉汤的小三角瓶中, 37 ℃水浴振荡培养10 h,分别取0.5 μL菌液穿刺于2份上述制备的Ep管中,于37 ℃生化培养箱中培养(1份用于测序,另1份用于保存),余下的菌液用碱裂解法抽提质粒DNA。
具体步骤如下:(1)菌液全部转入50 mL离心管中, 4 000 r/min室温离心10 min,弃上清。(2)向细菌沉淀加入100 μL冰水预冷的溶液Ⅰ[50 mM葡萄糖, 25 mM Tris-HCl(pH 8.0),10 mM [EDTA(pH 8.0)],于漩涡振荡器剧烈振荡,使沉淀迅速充分分散。(3)将分散后的菌液转入EB管中,加入200 μL新配制的溶液 Ⅱ (1%SDS, 0.2 mol/L NaOH),盖紧管口,温和颠倒数次。(4)加入150 μL用冰水预冷的溶液Ⅲ(5 mol/L乙酸钾60 mL,冰乙酸11.5 mL,水 28.5 mL),盖紧管口,温和颠倒数次至出现白色絮状沉淀后, 4 ℃ 12 000 r/min 离心10 min。(5)小心转移上清至另一Ep管中,加入等体积的异丙醇,室温静置10 min后, 4 ℃ 12 000 r/min离心10 min。(6)弃上清,加入70%乙醇700 μL洗涤DNA沉淀, 4 ℃ 12 000 r/min 离心5 min。(7)弃上清,自然干燥后加入含蒸馏水50~100 μL溶解质粒DNA。(8)加入 10 mg/mL的RNaseA 5 μL,置37 ℃恒温水浴箱消化30~60 min。(9)弃3 μL进行电泳,观察结果。余下的DNA则保存于-20 ℃冰箱中备用。
将1#~6#质粒DNA凝胶于80 V电泳50 min,初步鉴定为阳性的质粒DNA作进一步的鉴定,即酶切和PCR鉴定。
2.2.7 重组克隆载体阳性菌株的鉴定:选取6#阳性克隆质粒经限制性内切酶酶切、PCR扩增和测序方法进行鉴定。
(1)质粒PCR鉴定:
6#质粒DNA 2.0 μL
CDV-H-ExAs 2.0 μL
CDV-H-ExS 2.0 μL
2×TaqPCR Mastermix 12.5 μL
ddH2O 6.5 μL
将此反应体系按如下反应条件进行反应:95 ℃预变性5 min; 95 ℃变性30 s, 65 ℃ 复性30 s,72 ℃延伸90 s,进行35个循环,最后72 ℃延伸10 min。
(2)重组质粒的双酶切鉴定:根据设计的上下游引物所带的酶切位点,选用BamHⅠ和HindⅢ进行双酶切。反应体系为20 μL体系,即:
重组质粒 6.0 μL
10×K Buffer 2.0 μL
BamHⅠ 1.0 μL
HindⅢ 1.0 μL
ddH2O 10.0 μL
离心混合均匀,置37 ℃水浴消化2 h,1%琼脂糖凝胶电泳观察酶切结果。将鉴定为阳性的重组质粒记为pMD18-T-H。
2.3 原核表达载体的构建
2.3.1 目的基因与载体的连接:分别将pMD18-T-H和pET32a(+)空载体用BamHⅠ和HindⅢ双酶切,用凝胶回收试剂盒回收目的片段。
pMD18-T-H双酶切体系:
重组质粒DNA 35.0 μL
10×K Buffer 5.0 μL
BamHⅠ 1.5 μL
HindⅢ 1.5 μL
ddH2O 7.0 μL
pET32a(+)空载体双酶切体系:
pET32a(+)空载体 25.0 μL
10×K Buffer 5.0 μL
BamHⅠ 1.5 μL
HindⅢ 1.5 μL
ddH2O 17.0 μL
将上述酶切目的基因片段与表达载体进行连接。
连接体系:
10×T4 DNA Ligase Buffer 1.5 μL
酶切后的载体 2.0 μL
酶切后的目的基因 7.0 μL
T4 DNA Ligase 0.5 μL
ddH2O 4.0 μL
总体积15 μL, 16 ℃连接过夜。
2.3.2 连接产物的转化:将连接产物转化到 BL21(DE3) 感受态细胞,涂布于含氨苄青霉素100 μg/μL的LB平板上, 37 ℃培养20 h。挑取抗性菌落,以PCR和酶切方法鉴定阳性克隆。
2.3.3 阳性克隆的鉴定:主要按以下2种方法进行:
(1)质粒PCR鉴定:
质粒DNA 1.0 μL
CDV-H-ExAs 2.0 μL
CDV-H-ExS 2.0 μL
2×TaqPCR Mastermix 12.5 μL
ddH2O 7.5 μL
(2)双酶切方法鉴定:将PCR阳性菌液,提取重组质粒DNA,根据设计的上下游引物所带的酶切位点及表达载体上的酶切位点的位置,选择限制性内切酶BamHⅠ和HindⅢ进行酶切。
所加样品如下:
重组质粒 6.0 μL
10×K Buffer 2.0 μL
BamHⅠ 1.0 μL
HindⅢ 1.0 μL
ddH2O 10.0 μL
总反应体系为20 μL
以上各组分混匀,短暂离心后,于37 ℃温浴2 h,酶切产物经1.0%琼脂糖凝胶电泳,观察结果。
2.3.4 阳性克隆的序列测定:将经2.3.3鉴定阳性的质粒,穿刺后送大连宝生物工程有限公司测序。
3 结果
3.1 H基因扩增结果 以本实验室鉴定保存的pMD18-H克隆质粒为模板扩增H基因,所获得PCR产物经1%琼脂糖凝胶电泳,得大小约1 900 bp条带,与预期大小相符。见图1。
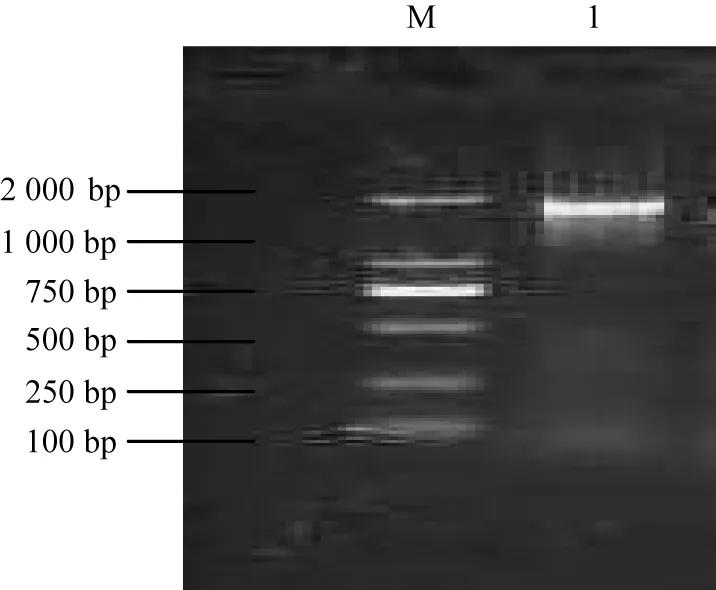
1:目的片段; M:DNA标准DL 2 000 图1 CDV GZ-Z株H基因的RT-PCR扩增结果
3.2 重组克隆载体的鉴定结果
3.2.1 PCR方法鉴定结果:将H基因的PCR产物与pMD 18-T载体连接构建重组质粒pMD18-H,再转化到感受态细胞E.coliDH5α进行蓝白斑筛选,挑取6个白色单个菌落接种于LB肉汤中培养,提取重组质粒DNA,取2 μL进行电泳,初步鉴定4#、 5#和6#为阳性。见图2。
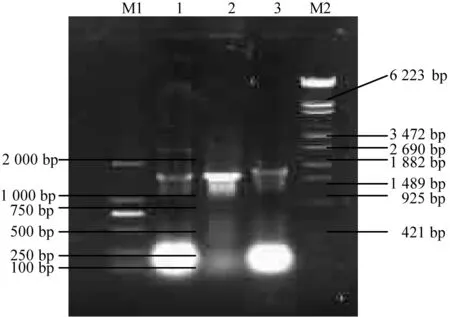
M1:DNA标准DL 2 000; M2:λ-EcoT14 Marker;1: 4#质粒PCR产物; 2: 5#质粒PCR产物; 3: 6#质粒PCR产物图2 4#、 5#、 6#重组质粒的PCR鉴定结果
3.2.2 双酶切方法鉴定结果:选取6#重组质粒用BamHⅠ和HindⅢ酶切鉴定,出现与预期结果相符的电泳带,表明重组质粒有外源基因片段的插入。并以6#重组质粒DNA为模板进行PCR反应并电泳观察,确定6#重组质粒为阳性克隆质粒。见图3。

M:λ-EcoT14 Marker;1:pET32a(+)双酶切; 2: 6#重组质粒双酶切 图3 6#重组质粒双酶切鉴定结果
3.3 重组表达质粒的鉴定结果
3.3.1 PCR方法鉴定结果:将重组质粒pET32-H转化入BL21(DE3)感受态细胞,涂布于含氨苄青霉素培养基上经筛选后。挑取抗性菌落进行PCR,PCR产物在1%琼脂糖凝胶电泳胶上呈单一条带。见图4。
3.3.2 酶切方法鉴定结果:选取1个PCR阳性重组子,提取质粒用BamHⅠ和HindⅢ 酶切鉴定,出现与预期结果相符的电泳带,表明重组质粒有外源基因片段的插入。见图5。
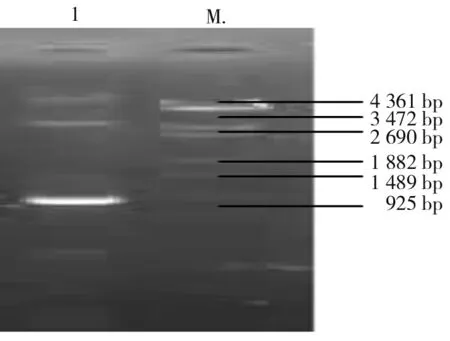
1:表达质粒PCR; M:λ-EcoT14 Marker 图4 PCR鉴定结果
3.3.3 测序鉴定结果:H基因重组质粒经琼脂糖凝胶电泳、质粒PCR及酶切鉴定为阳性后,穿刺保存2管,取其中1管送至大连宝生物公司测序,本实验选6#重组质粒进行测序,其中H基因ORF全长 1 842 bp,可编码607个氨基酸。具体序列长如下:
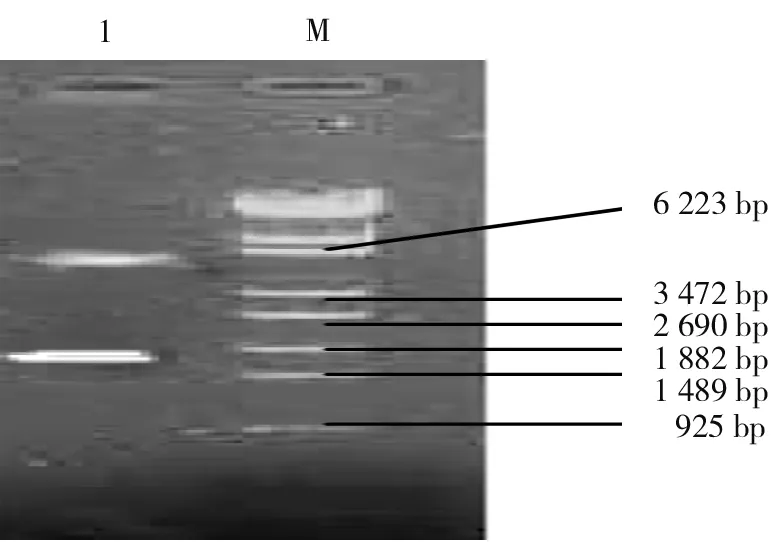
1:重组质粒双酶切;M:λ-EcoT14 Marker 图5 重组质粒双酶切的鉴定结果

4 讨论
4.1 犬瘟热的诊断方法 犬瘟热临床症状多样,主要表现为双相体温升高,急性鼻卡他和随后的支气管炎、卡他性肺炎、严重胃肠炎和神经症状等,而且容易继发其他细菌、病毒的混合感染和二次感染,靠传统的临床症状和流行病学资料只能作出初步诊断,要想准确快速地诊断犬瘟热较为困难,因此必须将传统方法与实验室诊断方法结合起来。目前,用于犬瘟热诊断的实验室方法有病原学检查、血清学检查、包涵体检查等。随着分子生物学研究的不断发展,分子生物学诊断技术,如核酸杂交技术和PCR技术,以敏感性高、特异性强而广泛应用于实验室和临床样品中病毒的检测。乔军和李金中等[17~19]分别报道了犬瘟热病毒RT-PCR诊断方法。孟庆龄等[20]用CDV犬冠状病毒(CCV)特异性引物对同一样品中CDV和CCV RNA模板进行联合RT-PCR扩增并对扩增条件进行了优化。PCR检测DNA的敏感性可以检测DNA的浓度来表示,而检测RNA的敏感性很难找到一个客观的指标。首先是不同组织或同一组织的不同时期特异性RNA的浓度不同;其次特异性RNA在细胞总RNA中的含量较低,而且还受RNA分离率和反转录率的影响;再者还受到所扩增片段在病毒复制过程中拷贝数的影响。用于诊断犬瘟热选择扩增片段以300~500 bp为宜。现在人们用分子生物技术对CDV 的检测进行了很多研究,其中PCR 技术尤以快速、灵敏、特异成为研究的重点。PCR技术以其敏感性高,特异性强而广泛用于病毒的检测。李金中[21]报道,根据Barrett报道的犬瘟热病毒Onderstepoort弱毒株的融合蛋白基因序列,设计合成了1对能扩增324 bp基因片断的引物,将提取的总RNA进行反转录,以此对引物进行PCR扩增,得到了与设计片断大小和酶切位点相同的产物,且不扩增犬细小病毒、犬腺病毒和狂犬病病毒3种病原的核酸,表现了PCR的特异性。同年,李金中[22]又利用套式PCR分段扩增了犬瘟热病毒融合蛋白的全基因。许莎琼等[23]报道,套式PCR 比RT-PCR 的检出率更高、更灵敏,可以作为对死前样品的有效检测手段。RT-PCR检测方法具有较高的灵敏度,与其它方法比较有许多优越性,相比病毒分离等方法,对实验室条件要求没那么高,而且快速方便,比较适合用于临床快速诊断,为CD的治疗提供可靠的依据,尤其可以在发病早期准确诊断,对及时更好地防治CD有很大的意义。迄今为止,病毒性疾病的诊断主要依赖免疫学方法、病毒分离培养和核酸分子杂交等,前两种方法敏感性较差,费时,假阴性较多;而用同位素标记的核酸探针作分子杂交则有放射性,操作繁杂,易污染;如用同位素标记则一般敏感性较差。PCR是病毒性疾病早期诊断的极有力工具,尤其是对难以进行病毒培养和血清学检验的病毒感染的诊断具有实用价值。
4.2 犬瘟热H蛋白基因 H蛋白基因由1 944~1 946 个核苷酸组成,它的mRNA的5’端有20个核苷酸的非编码区, 3’端非编码区至少含有100个核苷酸,在5’端或3’端非编码序列中没有发现稳定的茎环结构,仅有1个开放性阅读框架。 H蛋白是诱导机体产生中和抗体的主要蛋白之一,是抗CDV免疫的重要抗原。CDV通过H蛋白吸附到细胞表面的受体上,因此H蛋白决定CDV宿主的特异性,并协助F蛋白使CDV以囊膜与宿主细胞膜发生融合的方式进入宿主细胞,特别在启动细胞融合上起重要作用[24]。由此可见,H蛋白可用于CDV的免疫预防,且具有一定的诊断价值,其抗体可用于CDV感染的诊断或流行病学调查。据报道,H蛋白基因序列的系统发生分析表明CDV存在不同的基因型,新分离病毒H蛋白基因的遗传变异可能是近年来爆发犬瘟热的重要原因。本研究成功构建了H基因的原核表达载体,为CDV H基因的进一步研究打下了基础。CDV与MV和RPV的H蛋白的同源性分别为53%和52%,而MV和RPV的同源性为64%,三者总体同源性为36%,H蛋白同源性比较结果表明CDV从RPV分离出来远早于MV。H蛋白是诱导机体产生中和抗体的主要蛋白之一,是抗CDV免疫的很重要的抗原,H蛋白的变异率在麻疹病毒属所有结构蛋白中是最高的(按变异率从高到低依次为H、N、L、P、F、M蛋白),抗CDV H蛋白7个抗原决定簇中的6个McAb能中和CDV,阻止CDV的感染,但均不能封闭MV的感染及其血凝活性,而能抑制MV血凝活性并且中和MV的抗MVH蛋白的单抗也不能中和CDV,故H蛋白很可能在提议的新麻疹病毒属成员分类中起决定作用。
4.3 PCR反应 PCR的特异性一般取决于引物的特异性、退火温度、Mg2+浓度等因素,其中退火温度决定PCR能否高效率地扩增出特异性核酸片段。本试验以CDV-H-S和CDV-H-As为引物,在48 ℃ 退火进行扩增,能高效率获得特异性核苷酸片段,与理论计算的模板及引物退火温度基本相符。试验还对反应体系中Mg2+和dNTP浓度进行摸索,发现商品化带有Mg2+的PCR缓冲液及dNTP均可有效地扩增出目的片段。影响PCR结果的主要条件因素是MgCl2浓度和循环温度。
4.4 质粒DNA的提取 质粒DNA的提取是基因工程中的基本技术,目前常用的方法有碱变性法、羟基磷灰石柱层析法、质粒DNA释放法、酸酚法、两相法以及溴化乙锭-氯化铯密度梯度离心法,其中以碱变性法抽提质粒最常见。碱变性法提质粒的原理是根据染色体DNA和质粒DNA分子的大小、构型及变性与复性的差异而达到分离的目的。染色体DNA分子大,为双螺旋;质粒DNA分子小,为超螺旋。在pH 12.6的碱性条件下,染色体DNA的氢键断裂,但是螺旋共价闭合环状结构的2条互补链不完全分离。当以pH 4.8的醋酸钠高盐缓冲液调节pH到中性时,变性的质粒DNA可以恢复到原来的构型并保留在溶液中,而染色体DNA与不稳定的大分子RNA、蛋白质-SDS复合物等一起沉淀下来而被除去。
4.5 H基因的克隆与表达载体构建 本实验以扩增的H全基因片段为模板,分别设计引物通过PCR方法扩增部分缺失的H基因片段,将扩增的基因片段克隆到pMD18-T Simple vector中并测序。然后将H基因片段分别克隆入原核表达载体pET32a(+),构建了重组表达载体pET-32a-H,表达质粒经PCR和酶切鉴定。由于CDV是负链RNA病毒,因此在反转录中必须使用上游引物,而不是下游引物。
5 结论
5.1 本实验以实验室鉴定保存的pMD18-H克隆质粒为模板扩增H基因片段,并对其进行克隆,成功构建了克隆质粒pMD18-T-H。
5.2 将扩增得到的基因片段克隆入pET32a(+)载体中,成功构建了原核表达重组质粒pET32a-H。
[1] 夏咸柱.养犬大全[M].吉林:吉林人民出版社,1993.549~553.
[2] 蔡宝祥,殷震,谢三星,等.动物传染病诊断学[M].南京:江苏科学技术出版社,1993.4.
[3] 侯加法.小动物疾病学[M].北京:中国农业出版社,2005.77.
[4] 李金中,夏咸柱,邱薇,等.小熊猫等四种动物犬瘟热病毒的分离与鉴定[J].中国兽医学报,1998,28(8):8~10.
[5] 王兰萍.犬瘟热的诊断和防制研究进展[J].当代畜牧,2002,7(23):1.
[6] 殷震,刘景华.动物病毒学(第二版)[M].北京:科学出版社,1997.943~950.
[7] 莫小见,郭爱珍,陆承平,等.犬瘟热病毒南京株H蛋白基因的克隆与表达[J].中国病毒学,2004,19(5):487~489.
[8] 耿志贤,田克恭.犬瘟热病毒的分子生物学研究进展[J].中国兽医杂志,1997,23(2):45~46.
[9] 姜泊,张亚历,周殿元,等.分子生物学常用实验方法(第一版)[M].北京:人民军医出版社,1996.245~256.
[10] 乔军.犬瘟热病毒分子生物学特征研究进展[J].塔里木农垦大学学报,2002,14(1):39~44.
[11] 陆承平.兽医微生物学(第三版)[M].北京:中国农业出版社,2001.525~527.
[12] 杨应丽.犬瘟热病毒Lederle株H、N基因的克隆、序列分析及表达[D].北京:首都师范大学,2007.
[13] 王好.犬瘟热病毒H蛋白基因片段的克隆与原核表达[D].吉林:吉林农业大学,2007.
[14] 李建军,丁巧玲.我国犬瘟热研究进展[J].中国兽医杂志,2003,39(1):34~38.
[15] 孙园园,宫文妮,黄娟,等.犬瘟热病毒H基因克隆测序与原核表达[J].中国畜牧兽医,2009,36(5):58~62.
[16] J.萨姆布鲁克.分子克隆实验指南(第三版)[M].北京:科学出版社,2002.85~110.
[17] 乔军,孟庆龄,夏咸柱,等.犬瘟热病毒反转录聚合酶链反应检测方法的建立及初步应用[J].西北农业学报,2001,10(1):51~54.
[18] 乔军,孟庆龄,陈瑛,等.用半套式PCR检测犬瘟热病毒的研究[J].中国动物疫,2000,17(6):24~26.
[19] 李金中,何洪彬,夏咸柱,等.犬瘟热病毒反转录聚合酶链反应诊断方法建立和初步应用[J].病毒学报,1999,15(2):180~184.
[20] 孟庆龄,乔军,夏咸柱,等.犬瘟热犬冠状病毒联合RT-PCR检测方法的建立及应用[J].西南农业学报,2002,15(1):93~95.
[21] 李金中.犬瘟热病毒反转录-PCR诊断方法的建立和初步应用[J].病毒学报,1999,15(20):180~184.
[22] 李金中,夏咸柱,殷震.应用套式PCR分段扩增犬瘟热病毒融合蛋白的全基因[J].中国兽医学报,1999,12(2):124~125.
[23] 许莎琼,朱建国,傅志强,等.犬瘟热病毒当前研究进展[J].上海畜牧兽医通讯,2006,(1):5~7.
[24] Lamb R A.Paramyxovirus fusion:a bypothesis for changes[J].Virology,1993,197(20):1~11.
Constructing Prokaryotic Expression Vector of Canine Distemper Virus GZ-Z Strain of H Gene
Xue Lei
(Meitan Agricultural Bureau of Guizhou, Meitan Guizhou 564100,China)
In this experimental, previous experiments H construct containing canine distemper virus gene pMD18-T vector, according to pMD18-H sequences withBamHⅠ andHindⅢ restriction sites of the primers, PCR amplification of the H gene, are about 1 949 bp fragments around. The fragment was cloned into pMD18-Tsimple vector withBamHⅠ andHindⅢ restriction enzyme digestion were identified, plasmid PCR identification, and selected positive clones. The positive clones will then beBamHⅠandHindⅢ restriction enzyme digestion,purification of gene fragments recovered CDVH; the original expression vector pET32a(+) digested with the same method,recovery vector fragment with T4 ligase to connect to the recovery of the pET32a(+)vector to construct the prokaryotic expression plasmid pET32-H, after the byBamHⅠ andHindⅢ restriction enzyme digestion, plasmid PCR, sequencing series of identification. The results show that, pET32-H prokaryotic expression plasmid was successfully constructed, which insert size of 1 949 bp, encoding 648 amino acid residues. The experiment for the next H protein expression and lay the foundation for the preparation of monoclonal antibodies
Canine Distemper Virus; Constructing of Expression; PCR; H Gene

2015-08-19
S858.292:Q7
A
1007-1474(2015)06-0016-09
猜你喜欢
环球时报(2022-09-20)2022-09-20 15:18:57
现代畜牧科技(2021年7期)2021-07-28 06:40:42
今日农业(2020年24期)2020-12-15 16:16:00
山西医科大学学报(2017年11期)2017-12-01 07:22:37
食品研究与开发(2016年9期)2016-06-13 08:26:16
兽医导刊(2016年12期)2016-05-17 03:51:50
哈尔滨商业大学学报(自然科学版)(2016年1期)2016-04-22 07:04:42
农村百事通(2015年8期)2015-05-19 08:07:27
现代检验医学杂志(2015年4期)2015-02-06 02:02:06
养殖与饲料(2014年10期)2014-02-28 22:14:59
