
高效液相色谱法测定酒石酸泰乐菌素有关物质的研究
2015-06-09 22:01包洁华张金霞郭强功张萍
中国兽药杂志 2015年2期
包洁华,张金霞,郭强功,张萍
(宁夏泰瑞制药股份有限公司,银川750101)
高效液相色谱法测定酒石酸泰乐菌素有关物质的研究
包洁华,张金霞∗,郭强功,张萍
(宁夏泰瑞制药股份有限公司,银川750101)
依据欧洲药典讨论稿中酒石酸泰乐菌素有关物质的检测方法,在280 nm波长处,以pH 5.5的磷酸盐缓冲溶液-乙腈-水(10∶27.5∶62.5,V/V)为A流动相,以pH 5.5的磷酸盐缓冲溶液-水-乙腈(10∶40∶50,V/V)为B流动相进行梯度洗脱,并对专属性、准确度、精密度、检测限和定量限、线性和范围、耐用性、及溶液稳定性进行考察。结果显示,杂质A、N、O、R、S峰峰面积与其对应浓度呈线性关系,其相关系数r均大于0.999,各项考察指标均符合要求。结果证明该分析方法能科学、合理的分析、控制酒石酸泰乐菌素有关物质,可以持续、准确地反映酒石酸泰乐菌素的特性。
高效液相色谱法;酒石酸泰乐菌素;梯度洗脱;有关物质
酒石酸泰乐菌素是一种畜禽专用抗感染和促生长的抗生素[1],属大环内酯类,主要对多数革兰氏阳性菌、革兰氏阴性球菌、厌氧菌及军团菌、支原体及衣原体有良好效果[2]。广泛应用于鸡、牛、羊、猪的感染性疾病。《中华人民共和国兽药典》中利用液相色谱法测定酒石酸泰乐菌素组分[3],有关物质检测方法尚无报道,产品中有关物质定性不全面。然而,随着科技不断进步和国际市场的竞争日益剧烈,对兽用抗生素的质量已经不再单一要求高组分和高含量,对产品中的杂质和残留已经越来越受到广泛关注。由于抗生素发酵过程中,细菌培养液中代谢产物的组分非常复杂,使用一般的液相色谱法进行分析时,虽然主要组分可以在相对比较短的时间内分离完毕,但有一些杂质组分往往得不到充分洗脱。本文参考欧洲药典(EP)[4]和美国药典(USP)[5],研究并确立了梯度洗脱高效液相色谱法测定酒石酸泰乐菌素中的有关物质。
1 仪器与试剂
1.1 仪器 高效液相色谱仪(岛津LC-2010AHT)、CWA-VN3超纯水机(北京普析通用仪器有限责任公司)、BSA224s电子分析天平(德国赛多利斯)。
1.2 试剂
1.2.1 酒石酸泰乐菌素标准品(批号:FOD333,来源:美国药典委员会)、酒石酸泰乐菌素(批号:A91140702,由宁夏泰瑞制药股份有限公司提供)、磷酸氢二钾(分析纯)、磷酸二氢钾(分析纯)、乙腈(色谱纯)。
1.2.2 pH 5.5的磷酸盐缓冲溶液 取27.22 g磷酸二氢钾于适量水中溶解,并用水稀释至1000mL,再用34.84 g/L的磷酸氢二钾溶液调节pH值至5.5±0.1。
2 方法与结果
2.1 液相色谱条件 岛津LC-2010高效液相色谱仪,末端封尾C18色谱柱,规格4.6 mm×250 mm× 5μm,紫外检测器,波长280 nm,柱温60℃[4],流速1.0 mL/min,以pH 5.5的磷酸盐缓冲溶液-乙腈-水(10∶27.5∶62.5,V/V/V)为A流动相,以pH 5.5的磷酸盐缓冲溶液-水-乙腈(10∶40∶50,V/V/V)为B流动相,梯度洗脱[6]程序如表1。

表1 梯度洗脱流动相比例变化
2.2 专属性
2.2.1 空白溶液 流动相、水。
2.2.2 供试品溶液的制备 称取供试品25.0 mg,精密称定,置25 mL量瓶中,用流动相B溶解并定容至刻度,摇匀后用0.45μm滤球过滤。
2.2.3 对照品溶液的制备 对照溶液(a):精密称取5 mg酒石酸泰乐菌素标准品(包含泰乐菌素A,B,C和D组分,以及杂质A,N,O,R和S)用流动相B溶解并稀释至5.0mL,摇匀后用0.45μm滤球过滤。
对照溶液(b):准确移取1.0 mL供试品溶液置量瓶中,用水稀释至100.0mL,再取1.0mL该溶液用水稀释至10.0mL,摇匀后用0.45μm滤球过滤。
2.2.4 测定 精密量取空白溶液、对照品溶液与供试品溶液各20μL注入高效液相色谱仪,记录色谱图至泰乐菌素A组分保留时间的1.4倍(图1)。空白溶液对供试品杂质没有干扰,同时供试品中杂质峰的相对保留时间与标准品中杂质峰的相对保留时间一致(表2)。

图1 专属性考察色谱图
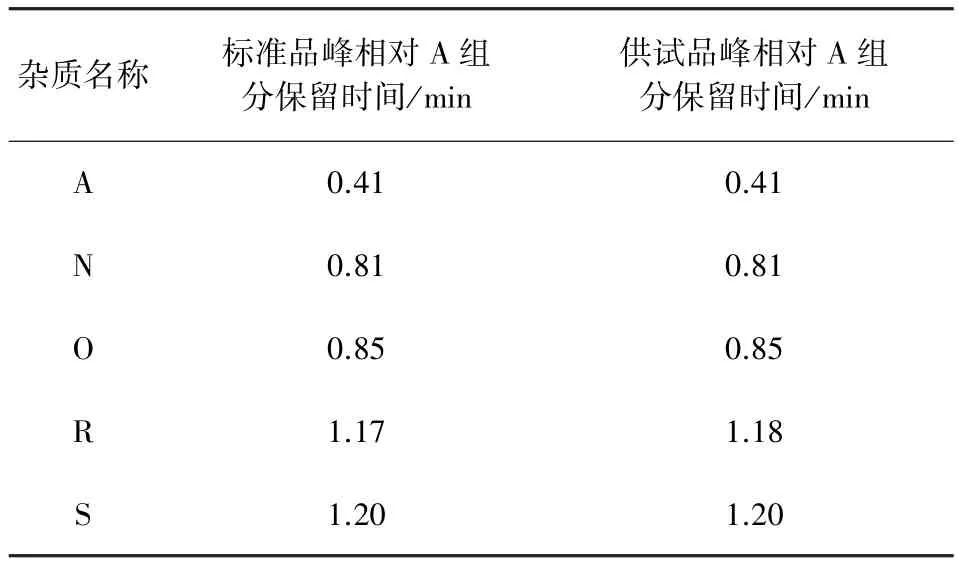
表2 专属性考察数据
2.3 准确度 称取酒石酸泰乐菌素供试品25.0mg置于50 mL量瓶中,用流动相B溶解并定容至刻度,精密量取20μL注入高效液相色谱仪,进样2次,记录色谱图至泰乐菌素A峰保留时间的1.4倍。分别称取酒石酸泰乐菌素标准品4、5、6mg置10mL量瓶中,用供试品溶液溶解并定容至刻度,相对浓度为80%、100%、120%。分别精密量取各溶液20μL注入高效液相色谱仪,各进样3次,记录色谱图至泰乐菌素A峰保留时间的1.4倍。然后计算三个浓度下(80%、100%、120%)测试杂质的面积,测试结果及计算回收率结果见表3。三个浓度下(80%、100%、120%)测试杂质A、N、O、R、S面积回收率均在80%~120%,相对标准偏差均小于10.0%。
2.4 精密度 分别称取酒石酸泰乐菌素供试品20.0、25.0、30.0mg,分别置于3个25mL量瓶中,用流动相B溶解,制备出浓度为80%、100%、120%的供试品溶液,同法制备3个浓度共9个供试品溶液,分别编号为1、2、3。精密量取各溶液20μL注入高效液相色谱仪,记录色谱图至泰乐菌素A峰保留时间的1.4倍。按峰面积归一化法计算三个浓度下(80%、100%、120%)测试选定杂质的允许差(即极差值),测试结果及计算结果见表4。三个浓度下(80%、100%、120%)测试选定杂质的允许差(即极差值)均不大于0.10%。

表3 供试品五种杂质A、N、O、R、S回收率结果

表4 五种杂质A、N、O、R、S重复性测试结果
2.5 检测限和定量限
2.5.1 供试品溶液制备 称取5 mg酒石酸泰乐菌素标准品用流动相B溶解并稀释至5.0 mL,作为标准品溶液1(可以使用当日系统适用性溶液),编号为1,依次2倍稀释至杂质A,N,O,R和S中最大杂质信噪比接近10∶1及3∶1(稀释溶液依次编号为2、3、4…n),备用。
2.5.2 精密量取空白溶液、标准品溶液(1、2、3、4…n)各20μL注入高效液相色谱仪,各进样1次,记录色谱图。分别计算色谱图中对应峰信号响应值与空白溶液信号响应值的比值。计算以杂质A、N信噪比约为3∶1时的溶液浓度为0.03125 mg/m L即为检测限,杂质O、R、S信噪比约为3∶1时的溶液浓度为0.06250 mg/mL即为检测限,杂质A、O信噪比约为10∶1时的溶液浓度0.25 mg/m L即为定量限,杂质N、R、S信噪比约为10∶1时的溶液浓度0.125 mg/mL即为定量限。
2.6 线性和范围 称取酒石酸泰乐菌素标准品60.0 mg,精密称定,置50 mL容量瓶中,浓度为1.2 mg/mL,作为供试品溶液。分别吸取供试品溶液1.0、7.9、5.8、4.6、2.1、1.0 mL,配制成1.2、0.95、0.7、0.55、0.25、0.125 mg/mL浓度的线性溶液,按系统色谱条件检测,记录各杂质峰的峰面积。以杂质峰的峰面积对应供试品浓度做回归曲线,数据及计算结果见表5。

表5 五种杂质A、N、O、R、S的线性曲线数据结果
2.7 耐用性和溶液稳定性考察 通过调整流动相流速1.00±0.05 mL/min、波长280±2 nm、pH值5.5±0.1,对杂质A、N、O、R、S进行检查,结果的允许差均不大于0.10%。说明微小改变色谱条件适用于这五种杂质的检测。对溶液稳定性进行考察,标准品溶液、供试品溶液分别在常温放置0、12、24、48、60、72 h,取临用新配标准品溶液进行检测。测定的结果允许差(即极差值)均不大于0.10%。
3 讨论与小结
本文参照欧洲药典讨论稿中酒石酸泰乐菌素有关物质检测方法,讨论稿中包括的杂质有A、N、O、R、S、E、G、H、I、J、Q共11种,本文中只对A、N、O、R、S进行了考察,其余杂质有待进一步考察。
欧洲药典讨论稿中试样溶解用乙腈和水,梯度洗脱程序是0~25 min,流动相A 100%洗脱,25~45 min流动相A为100→84,流动相B为0→16,45~65 min时流动相A为84%,B为16%保持恒定,洗脱过程中色谱峰杂质的分离度达不到要求[3](R≥1.5),调整梯度洗脱程序为0~20 min,流动相A 100%洗脱,20~45 min流动相A为100→84,溶剂调整为流动相B后色谱分离效果良好,不会产生因为溶剂与流动相不一致或者是相溶性差而导致的样品析出再二次溶解的问题,避免出现色谱峰对称性差甚至分叉的问题。
试验中对室温、30、40、50℃等条件进行了考察,色谱图出峰时间延长,且柱压明显升高。另外,方法中对耐用性进行考察,包括调整流速1.00±0.05 mL/min、波长280±2 nm、pH值5.5± 0.1,结果的允许差均不大于0.10%。说明微小改变色谱条件适用于酒石酸泰乐菌素有关物质A、N、O、R、S的检测。
酒石酸泰乐菌素的生产中除A、B、C、D组分外,杂质组分多,采用梯度洗脱能使杂质组分得到强有力的分离。
关于酒石酸泰乐菌素有关物质检测方法目前没有相关报道,本文建立了酒石酸泰乐菌素五种杂质A、N、O、R、S的检测方法。五种杂质A、N、O、R、S在测定中均能获得良好的线性曲线,该方法专属性、准确度好,对更好地控制产品质量具有一定的实用价值。
[1] 酒石酸泰乐菌素[Z].农村养殖技术,2012,(9).
[2] 李树纲,于清伟,张蕾,等.浊度法测定酒石酸泰乐菌素可溶性粉效价研究[J].中兽医医药杂志,2012,31(2):33-35.
[3] 中国兽药典委员会.中华人民共和国兽药典二○一○年版(二部)[S].
[4] The Directorate for the Quality of Medicines&Health Care of the Council of Europe(EDQM).European Pharmacopoeia[S].
[5] The United States Pharmacopeial Convention.U.S.Pharmacopoeia,37th Revision[S].
[6] 张玉奎,王杰,张维冰,等.实用高效液相色谱法的建立[M]北京:华文出版社;2001:413-414.
(编辑:陈希)
Determ ination of Tylosin Tartrate Related Substances by HPLC
BAO Jie-hua,ZHANG Jin-xia∗,GUO Qiang-gong,ZHANG Ping
(Ningxia TaiRui Pharmaceutical Co.,Ltd,Yinchuan 750101,China)
The determination method of tylosin tartrate related substances from EP discussion draft,At the wavelength of 280 nm,pH 5.5 phosphate buffer solution-acetonitrile-water(10∶27.5∶62.5,V/V)as the mobile phase A,pH 5.5 phosphate buffer solution-water-acetonitrile(10∶40∶50,V/V)as themobile phase B gradient elution,and the specificity,accuracy,precision,limit of detection and limit of quantification,linearity and range,durability,and solution stability was investigated.The results showed that the impurity A,N,O,R,S peak area and the corresponding concentration showed a linear relationship,the correlation coefficient r was greater than 0.999,the indicators are in line with inspection requirements.It was demonstrated that the method can scientific,reasonable analysis,control of tylosin tartrate related substances,It can be sustained,accurately reflect the characteristics of tylosin tartrate.
HPLC;tylosin tartrate;gradient elution;relevance substance
2014-10-29
A
1002-1280(2015)02-0035-05
S859.796
包洁华,从事抗生素生产质量管理工作。
张金霞。E-mail:371226727@qq.com
猜你喜欢
天津化工(2022年2期)2022-04-26
化工生产与技术(2021年3期)2021-07-08
食品与生物技术学报(2020年4期)2020-01-06
江苏农业科学(2019年5期)2019-09-02
中成药(2017年4期)2017-05-17
中国药房(2017年6期)2017-03-29
人民周刊(2016年11期)2016-06-30
中国感染与化疗杂志(2015年5期)2015-01-23
中国药业(2014年21期)2014-05-26
食品工业科技(2014年13期)2014-03-11
