
Determ ination of Levam isole Hydrochloride in Levam isole Hydrochloride Powder by HPLC
2015-06-09 22:01:14WANGLi,LIUSu-mei,ZANGHe-ying等
中国兽药杂志 2015年2期
Determ ination of Levam isole Hydrochloride in Levam isole Hydrochloride Powder by HPLC
Themethod for determination of levamisole hydrochloride in levamisole hydrochloride powder by HPLC was developed.The analyte was separated on C18 chromatographic column using a mobile phase of potassium dihydrogen phosphate(0.05 mol/L,pH value adjusted to 7.0 with triethylamine)-acetonitrile(80∶20),and detected at the wavelength of 214 nm.Themethod was validated for specificity,accuracy,linearity,precision and robustness.The results showed that a good linear relationship was accepted in the range of 0.1~2.0 mg/mL(R2=0.9995),and the average recovery was 101.3%(n=6),and the RSD value was 0.6%.Thismethod was suited to control the quality of levamisole hydrochloride powder.
levamisole hydrochloride powder;levamisole hydrochloride;HPLC
盐酸左旋咪唑为广谱驱虫药,对牛、羊、猪、犬和鸡的大多数胃肠道线虫具有良好驱虫活性。兽药中有盐酸左旋咪唑片、盐酸左旋咪唑注射液和盐酸左旋咪唑粉等不同剂型,其中盐酸左旋咪唑粉收载于《兽药国家标准汇编—兽药地方标准上升国家标准》第二册[1]。标准中采用三氯甲烷萃取、高氯酸滴定法进行含量测定。检测中试剂三氯甲烷对人体有危害,对环境污染也较严重,且操作步骤较繁琐;同时高氯酸滴定液能与多种药物反应,专属性不高。目前相关文献[2-6]报道测定盐酸左旋咪唑含量的方法除高氯酸滴定法外,还有紫外分光光度法和高效液相色谱法。其中紫外分光光度法也易受其他药物的影响,专属性较差。刘素梅等[2]、曾建亭等[3]和陆兴毅[6]分别报道采用HPLC法测定万乳康、盐酸左旋咪唑片和盐酸左旋咪唑糖浆等制剂中盐酸左旋咪唑的含量,但目前尚未有高效液相法测定盐酸左旋咪唑粉中盐酸左旋咪唑含量的报道。
本研究采用高效液相色潽法测定盐酸左旋咪唑粉中盐酸左旋咪唑的含量,试验结果表明其方法简便、准确可靠、重现性好,能够有效的控制该产品的质量。
1 仪器和试药
1.1 仪器与设备 Waters e2695高效液相色谱仪,配紫外检测器;METILER XP205电子天平。
1.2 试药及试剂 盐酸左旋咪唑对照品纯度为99.9%,批号100167-201203,购于中国食品药品检定研究院;盐酸左旋咪唑粉(规格:5%)为市场购买;乙腈为色谱纯试剂,水为超纯水,其他试剂为分析纯。
2 方法与结果
2.1 检测波长的选择 精密称取盐酸左旋咪唑对照品适量,用水稀释制成每1 mL含5μg的溶液,在200~300 nm波长范围内进行光谱扫描,得出214 nm为盐酸左旋咪唑最大吸收波长,所以选择214 nm作为该方法的测定波长。结果见图1。
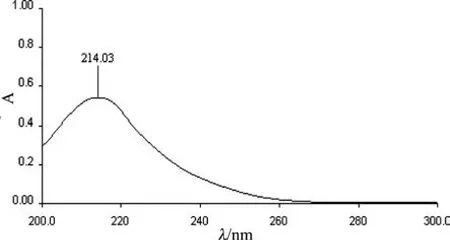
图1 盐酸左旋咪唑的紫外光谱图
2.2 色谱条件 Waters X-Bridge C18(5μm,4.6 mm×150 mm)色谱柱,流动相为0.05 mol/L磷酸二氢钾(用三乙胺调节pH值为7)-乙腈(80∶20);流速为1.0 mL/min;检测波长为214 nm;柱温为室温;进样量为10μL。
2.3 溶液的制备
2.3.1 对照品溶液的制备 精密称取盐酸左旋咪唑对照品100 mg,置50 mL量瓶中,加水溶解稀释至刻度,摇匀,配制成2 mg/mL的储备液。精密量取适量,用水稀释成0.5 mg/mL的对照品溶液,即得。
2.3.2 供试品溶液的制备 取样品适量(约相当于盐酸左旋咪唑50mg),置100mL量瓶中,用水溶解稀释至刻度,摇匀,即得。
2.3.3 阴性对照溶液的制备 取按照处方比例制备的不含盐酸左旋咪唑的阴性样品,按2.3.2项下方法制成阴性样品溶液。
2.4 线性关系考察 取2.3.1项对照品储备液,用水稀释成浓度为0.1、0.2、0.5、1.0、2.0 mg/m L的系列对照品溶液,按2.1项下方法测定,记录色谱图,以对照品溶液浓度为横坐标,相对对应的峰面积为纵坐标,进行线性回归,回归方程为:Y=23598708X+634363,相关系数R2=0.9995,即在0.1~2.0 mg/mL范围内,盐酸左旋咪唑浓度与峰面积呈现良好的线性关系。结果见图2。
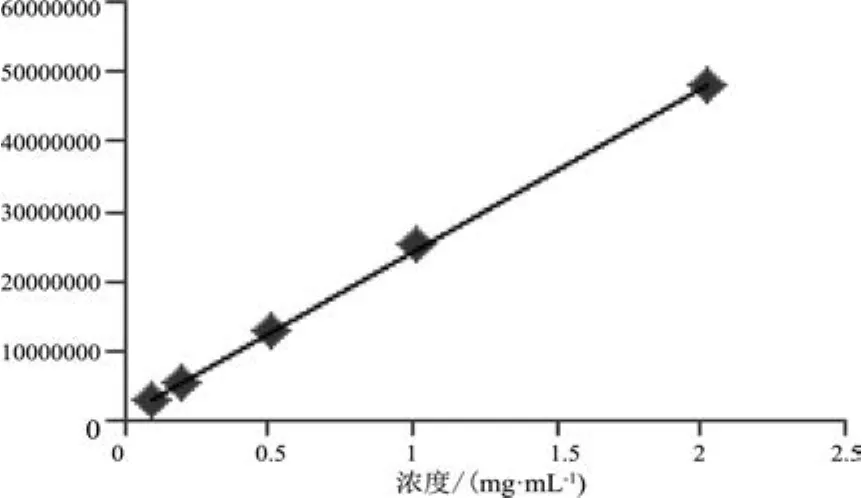
图2 盐酸左旋咪唑标准曲线图
2.5 专属性 取对照品溶液、阴性对照溶液,按2.1项下方法测定,记录色谱图,结果表明阴性无干扰,结果见图3、图4。
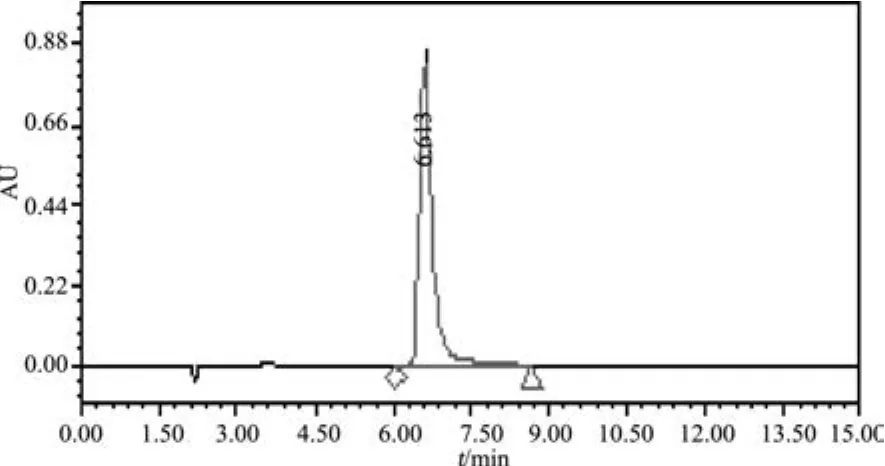
图3 盐酸左旋咪唑对照(0.5mg/m L)色谱图

图4 阴性样品色谱图
2.6 耐用性
2.6.1 不同品牌和型号的色谱柱 分别选用Agilent ZORBAX SB-C18(5μm,3.0mm×250mm)、Agela Venusil MP C18(5μm,4.6 mm×250 mm)、Waters X-Bridge C18(5μm,4.6 mm×150 mm),按2.1项下方法测定,记录色谱图,结果见表1。

表1 耐用性试验结果表(不同色谱柱)
2.6.2 不同比例流动相 流动相0.05mol/L磷酸二氢钾(用三乙胺调节pH值为7.0)-乙腈分别以不同比例,按2.1项下方法测定,记录色谱图,结果见表2。

表2 耐用性试验结果表(不同比例流动相)
2.6.3 不同pH值 将流动相0.05 mol/L磷酸二氢钾-乙腈(80∶20)分别用三乙胺调节0.05 mol/L磷酸二氢钾的pH值为6.8、7.0、7.2,按2.1项下方法测定,记录色谱图,结果见表3。
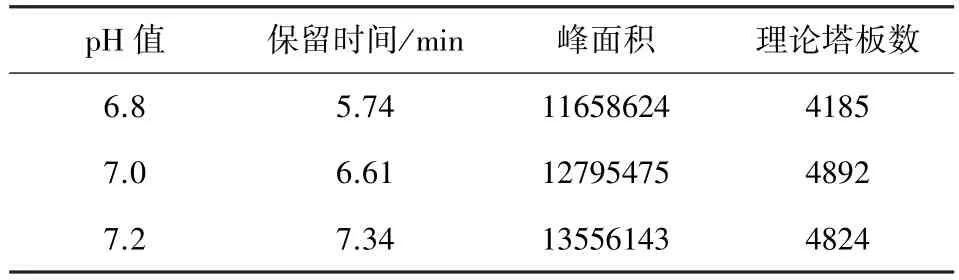
表3 耐用性试验结果表(流动相不同pH值)
结果表明,分别在不同品牌和型号色谱柱、不同比例流动相和不同pH值流动相的条件下,各试验结果虽略有不同,但均能满足试验需要。
2.7 准确度与精密度 取同一批号已知含量的盐酸左旋咪唑粉(47.58 mg/g)0.5 g,精密称定,共6份,分别加入盐酸左旋咪唑对照品适量,按2.3.2项下方法处理样品后,记录色谱图,外标法以峰面积计算盐酸左旋咪唑的含量,结果表明,该方法回收率高,且重复性好。结果见表4。
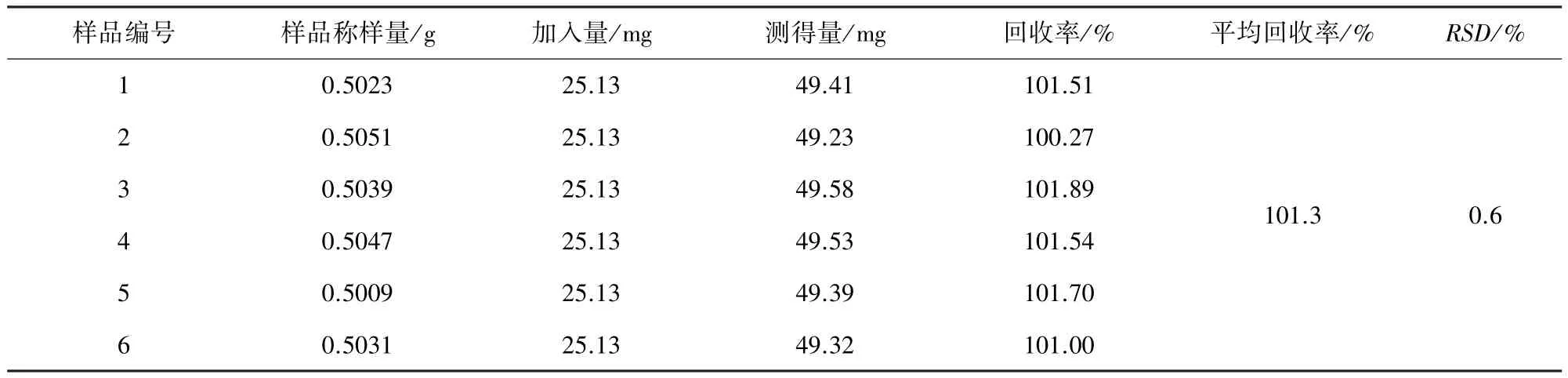
表4 回收率试验结果表
2.8 稳定性 取精密度试验项下的一个样品溶液,分别在0、2、4、8、12 h按2.1项下方法测定,记录色谱图,考察样品配制溶液的稳定性,结果表明样品溶液在12 h内稳定。结果见表5。

表5 稳定性试验结果表
2.9 样品测定 取5批样品,按2.3.2项下方法处理样品后,按2.1项下方法测定,记录色谱图,外标法以峰面积计算盐酸左旋咪唑的含量,并将结果与非水滴定法测定结果比较,结果表明,两种方法测定结果基本一致。结果见表6。
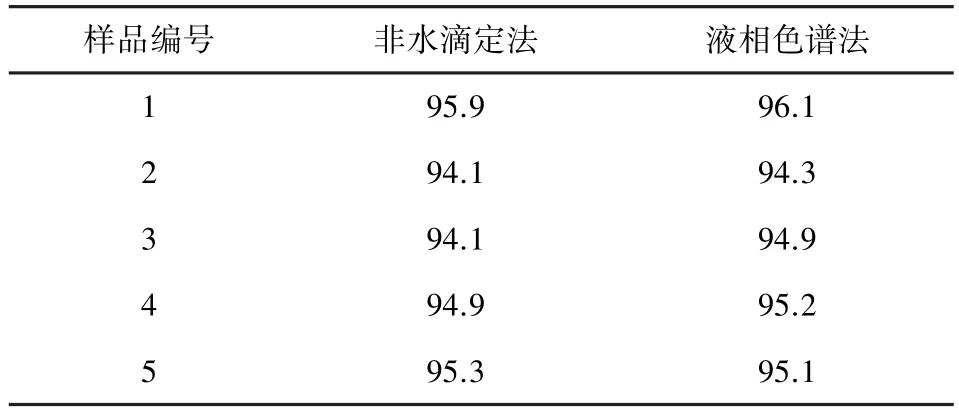
表6 样品测定结果表(标示量/%,n=5)
3 讨论与小结
原标准规定的高氯酸滴定法,要求试验条件比较苛刻,滴定时必须在非水条件下进行。试验中需用三氯甲烷进行萃取,不但操作繁琐而且该试剂毒性大,同时萃取时间不同,力度不同,对试验结果均有一定的影响。弱碱性物质及盐类也均可以与高氯酸反应,所以专属性不高。在高氯酸滴定法检测过程中还发现,该方法对辅料要求也很严格。当使用淀粉为辅料时,样品加水溶解再加入氢氧化钠试液后,短时间内就易产生糊化现象,使三氯甲烷不能完全萃取而影响含量测定的结果。
本研究采用水为溶剂,不但减少了有毒试剂的使用,操作简便,专属性强,而且也不受常用辅料的影响。试验结果显示高效液相色谱法与高氯酸滴定法相比,测定结果基本一致,并且方法简便、准确可靠、重现性好。高效液相色谱法可以用于盐酸左旋咪唑粉中盐酸左旋咪唑的含量测定。
[1] 兽药国家标准汇编—兽药地方标准上升国家标准第二册[S].
[2] 刘素梅,李华岑,韩立,等.万乳康中盐酸左旋咪唑含量测定方法的研究[J].中国兽药杂志,2010,44(2):26-28.
[3] 曾建亭,钟小燕.HPLC法测定盐酸左旋咪唑片的含量及溶出度[J].中国热带医学,2010,10(5):628-629.
[4] 徐灿辉,何维为.高效液相色谱法测定盐酸左旋咪唑片的含量[J].药物鉴定,2007,16(7):12-13.
[5] 王勇忠.高效液相色谱法测定盐酸左旋咪唑搽剂的含量[J].海峡药学,2007,19(12):52-53.
[6] 陆兴毅.HPLC法测定盐酸左旋咪唑糖浆的含量[J].药物分析杂志,2008,28(10):1753-1754.
(编辑:侯向辉)
WANG Li1,LIU Su-mei1,ZANG He-ying1,ZHAO Chen-yu2,HAN Li1,QIU Tian-bao1
(1.Henan Province Institute of Veterinary Drug and Feed Control,Zhengzhou 450008,China;2.Chongqing Medical University,Chongqing 400016,China)
猜你喜欢
中国民间疗法(2021年17期)2021-11-04 08:39:44
家庭医学(下半月)(2020年3期)2020-05-30 12:42:12
家庭医学(下半月)(2019年8期)2019-09-25 09:02:08
中国外汇(2019年10期)2019-08-27 01:58:22
中国外汇(2019年10期)2019-08-27 01:58:22
布达拉(2019年3期)2019-06-11 05:34:00
中国外汇(2019年22期)2019-05-21 03:15:02
中国外汇(2019年21期)2019-05-21 03:04:22
兽医导刊(2016年6期)2016-05-17 03:50:16
中国卫生标准管理(2015年2期)2016-01-14 03:41:34
