
羧苄西林钠有关物质的测定方法优化
2015-06-07 05:51朱佳莉
机电信息 2015年2期
朱佳莉
羧苄西林钠有关物质的测定方法优化
朱佳莉
(上海新亚药业有限公司新先锋药厂,上海201203)
对用现有国家标准中羧苄西林钠有关物质的方法测定过程中可能出现的问题进行了分析,并对原有的测定方法进行了优化,以达到更好的分离效果,使得有关物质间的分离明显改善。
羧苄西林钠;有关物质;液相色谱;方法优化
0 引言
羧苄西林钠为广谱青霉素类抗生素,通过抑制细菌细胞壁合成发挥杀菌作用。对大肠埃希菌、变形杆菌属、肠杆菌属、枸橼酸菌属、沙门菌属和志贺菌属等肠杆菌科细菌,以及铜绿假单胞菌、流感嗜血杆菌、奈瑟菌等其他革兰阴性菌具有抗菌作用。羧苄西林钠主要适用于系统性铜绿假单胞菌感染,如败血症、尿路感染、呼吸道感染、腹腔、盆腔感染以及皮肤、软组织感染等,也可用于其他敏感肠杆菌科细菌引起的系统性感染。
有关物质是指在生产过程中带入的起始原料、中间体、聚合体、副反应产物,以及储藏过程中的分解产物等。有关物质研究是药品质量研究中关键性的项目之一,其含量是反映药品纯度的直接指标。
高效液相色谱法(HPLC)是一种现代液相色谱法,其工作原理:用高压输液泵将流动相泵入到装有填充剂的色谱柱,注入的供试品被流动相带入柱内进行分离后,各成分先后进入检测器,数据处理装置对各种测量数据进行记录和处理,完成定性定量分析工作。由于高效液相色谱法采用了高压流动相、高效固定相和高灵敏度检测器,与其他色谱法相比,其具有分离效能高、选择性好、灵敏度高、分析速度快、适用范围宽等特点,已成为医药研究的有力工具。
1 用现行标准测定可能发生的问题
羧苄西林钠,英文名Carbenicillin Sodium for Injection,化学名为[6S-(2a,5a,6b)]-[(羧苯基-乙基)氨基]-3,3-二甲基-7-氧代-4-硫杂-1-氮杂双环[3.2.0]庚烷-2-甲酸二钠盐。
分子式:C17H16N2Na2O6S。分子量:422.4。
1.1 现行标准测定要求
现行标准为国家食品药品管理局起草的药品标准(WS1-C2-0042-89-2011),按该方法测定羧苄西林钠有关物质,其过程如下:
1.1.1 仪器与试药
仪器:Aglient 1260,HPLC主要组成参数如表1所示。
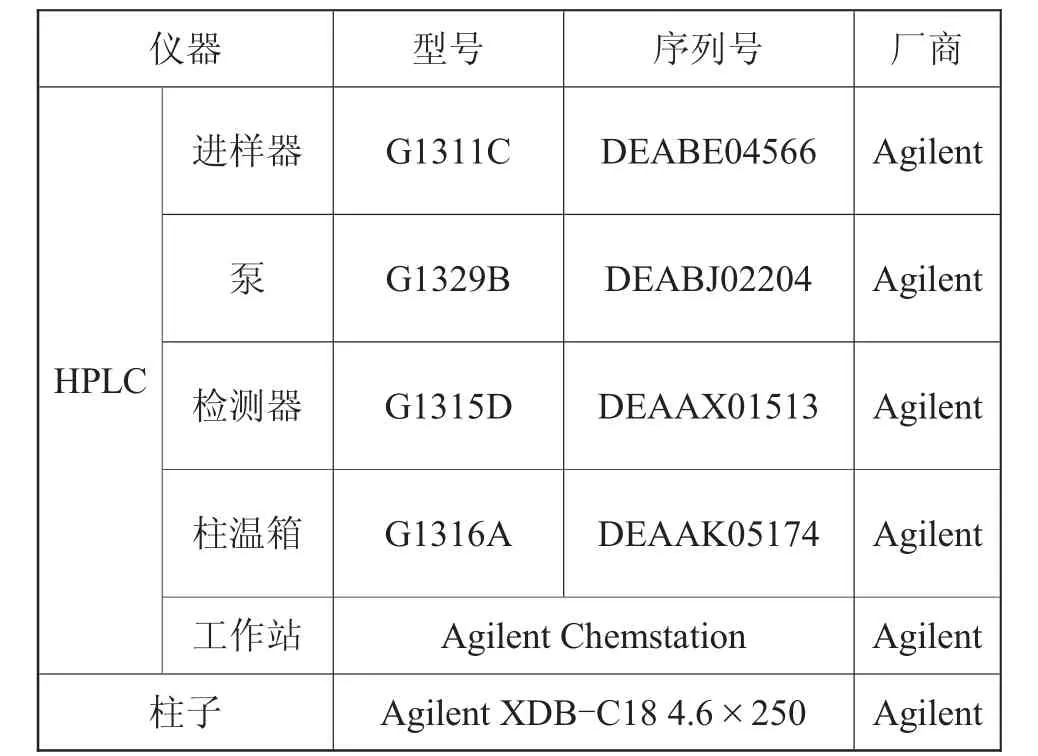
表1 Aglient 1260 HPLC主要组成参数
羧苄西林钠有关物质测定试药如表2所示。
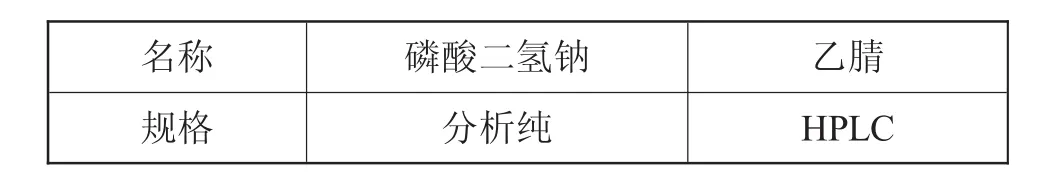
表2 羧苄西林钠有关物质测定所用试药
1.1.2 色谱条件
色谱柱:Agilent XDB-C18 4.6×250;波长:230 nm;流速:1m L/m in。
溶剂:取磷酸二氢钠7.8 g,加水溶解并稀释至900m L,用1moL/L氢氧化钠溶液调节pH值至6.4,用水稀释至1 000m L。
供试品配制:精密称定,加溶剂溶解并稀释成1m L中含3mg的溶液。
对照品配制:取青霉素对照品,精密称定,加溶剂溶解并定量稀释成1m L中含28μg的溶液。
计算:青霉素以无水物计,按外标法以峰面积计算。
流动相A:0.1moL/L磷酸二氢钠溶液(调节pH值至4.3)-乙腈(98∶2)。
流动相B:乙腈。
梯度洗脱程序如表3所示。

表3 梯度洗脱程序
1.2 测定结果
以羧苄西林钠有关物质测定结果中杂质个数最多的3批原料产品为例,其测定图谱分别如图1、图2、图3所示。

图1 测定结果图谱之1
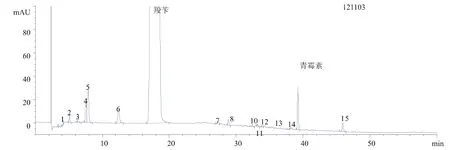
图2 测定结果图谱之2

图3 测定结果图谱之3
1.3 结果讨论
通过对本厂生产的65批羧苄西林钠产品进行有关物质的测定。其中,原料32批,对应制剂34批。用此梯度洗脱方法,有一些杂质峰未能完全分离。其中,杂质4和5的分离度非常不好。
针对羧苄西林钠有不良反应的投诉,需要对羧4.3,pH值过低,超出缓冲的缓冲范围。该溶液原有pH值为4.4,直接使用,不调节,杂质分离效果优化。
2.1.2 色谱柱选择
试用不同品牌色谱柱,有的出峰时间过早,有的分离效果不好。图4、图5为某不同品牌色谱柱出峰时间过早与分离效果不好的图谱。苄西林钠的有关物质检测方法进行优化,并对部分已知杂质进行定位,以求建立杂质图谱档案,更好地对羧苄西林钠进行质量监控。
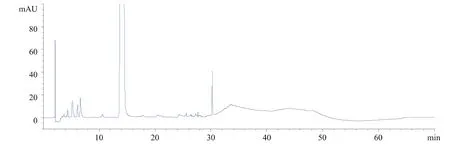
图4 某一品牌色谱柱出峰时间过早与分离效果不好的图谱
2 测定方法优化
2.1 色谱条件
2.1.1 流动相选择
流动相B为磷酸盐溶液,原标准中规定pH值为
考虑标准规定主峰保留时间约为20min,结合各杂质的分离情况,对比大量图谱后,最终选择了Sapphire C18色谱柱。
2.1.3 梯度洗脱程序的优化
经多次试验,现将梯度洗脱程序进行优化,其参数如表4所示。
2.1.4 其他色谱条件
仪器与试药、供试品的配制、对照品的配制均与原法相同。

图5 另一品牌色谱柱出峰时间过早与分离效果不好的图谱

表4 梯度洗脱程序的优化
2.2 溶液稳定性考察
当样品放置10m in,或者样品进样时温度过高,会加速杂质的生成。其中,杂质3明显增大,杂质15则消失,其图谱如图6所示。
2.3 优化测定结果
(1)对几个已知杂质分别进行定位。
(2)测定3批杂质最多的产品图谱,并重新进行定位。图7为对照品图谱,图8、图9、图10为供试品图谱。
(3)测定31批新生产的羧苄西林钠样品,测试结果显示方法稳定。
2.4 系统适应性试验
2.4.1 精密度试验
以对照品青霉素计RSD%=1.44%。
2.4.2 其他系统参数

图6 加速杂质的生成的图谱

图7 对照品图谱
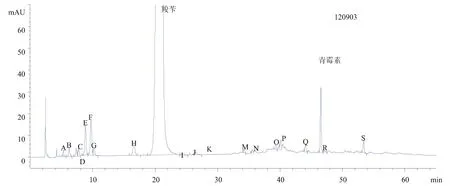
图8 供试品图谱1
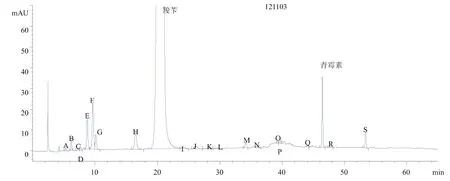
图9 供试品图谱2
以130202批号为例,所有塔板数均大于4 000,分离度除D、G外均符合要求,D、G虽未达到1.5的要求,但是也已经实现基本分离。
2.4.3 空白测试
空白测试图谱如图11所示。
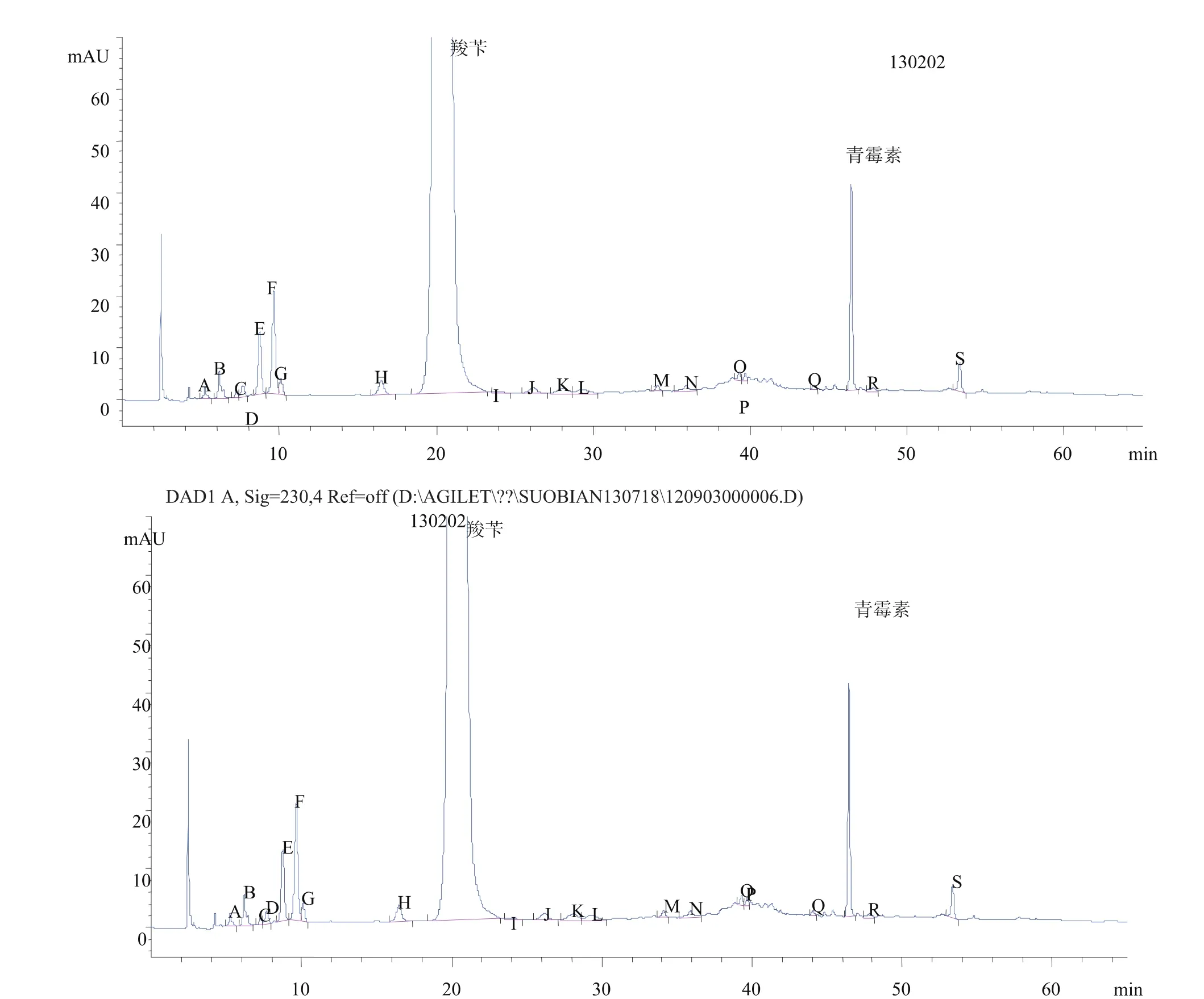
图10 供试品图谱3
图11 空白测试图谱
2.5 样品测定结果比较
用优化后的梯度洗脱方式测定,杂质F和G(即原来的杂质4和5)分离效果明显改善。全部杂质能够多定位出3个杂质峰。
3 结语
对现有国家标准中羧苄西林钠有关物质的测定方法进行优化,以达到更好的分离效果。因此,该优化方法可用于羧苄西林钠产品有关物质的跟踪测定。具体优化结论:
(1)流动相的配制:原标准中流动相A中混合了乙腈,为使溶剂更稳定,采用溶剂与有机相分开,仪器自动混合的方式;磷酸二氢钠溶液配制时,应尽量精密称定,保持流动相的统一性;配制流动相时,不用调节pH值;流动相在每次做样品时,要新鲜配制。
(2)供试品溶液:由于羧苄西林钠溶液分解速度很快,供试品配制时要临用新配;放置时间不宜超过5m in。
(3)色谱图记录时间:为达到杂质分离度要求,使所有杂质全部洗脱,总洗脱时间增加至80min。
(4)色谱柱选择:Sapphire C18色谱柱。
(5)有关物质中明显的峰的编号:标出A~S及青霉素共20个峰。
(6)对一些已知杂质的定位结果:杂质6-APA,未检出(出峰时间在A峰之前);杂质苯丙二酸为F峰;杂质D为4个物质的混合物,分别为C、D、E、F峰;杂质苯乙酸为O峰;杂质G为2个物质的混合物,分别为N、O峰。
2014-12-23
朱佳莉(1980—),女,上海人,助理工程师,研究对象:有关物质的液相色谱分析。
猜你喜欢
Chinese Physics B(2022年5期)2022-05-16
少先队活动(2020年12期)2021-01-14
艺术品鉴(2020年6期)2020-12-06
东坡赤壁诗词(2020年3期)2020-07-04
启迪与智慧·下旬刊(2019年2期)2019-09-10
中成药(2017年3期)2017-05-17
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
新丝路(下旬)(2016年10期)2016-06-05
领导科学论坛(2016年9期)2016-06-05
