
有机化合物生物富集因子的计算机预测研究
2015-06-05 09:51孙露陈英杰吴曾睿李卫华刘桂霞PhilipLee唐赟
生态毒理学报 2015年2期
孙露,陈英杰,吴曾睿,李卫华,刘桂霞,Philip W. Lee,唐赟
华东理工大学药学院 上海市新药设计重点实验室,上海 200237
有机化合物生物富集因子的计算机预测研究
孙露,陈英杰,吴曾睿,李卫华,刘桂霞,Philip W. Lee,唐赟*
华东理工大学药学院 上海市新药设计重点实验室,上海 200237
有机化合物在生物体内的富集,通常用生物富集因子(bioconcentration factor,简称BCF)来表达,这是化合物生态环境毒性评估的重要指标。为合理预测有机化合物是否易于生物富集,首先从美国环保局网站收集了624个具有不同BCF值的化合物,然后采用7种分子指纹结合5种机器学习方法(包括支持向量机、C4.5决策树、k最近邻法、随机森林法和朴素贝叶斯法),构建了化合物BCF的分类预测模型,所有模型均采用独立外部验证集进行验证。其中,使用ChemoTyper分子指纹结合支持向量机方法得到的二分类模型,整体预测准确度最好,达到了85.4%。通过采用信息增益、频率分析等方法,进一步确定了化合物中易于引起生物富集的关键子结构,包括芳基氯、二芳基醚、氯代烷烃等。研究中所用到的方法为有毒化学品的生态风险评价提供了良好可靠的预测工具。
生物富集因子;计算机预测;二分类模型;警示子结构;环境毒理学
生物从周围环境中吸收并累积某种元素或难分解的化合物,从而导致生物体内该物质的浓度超过环境中浓度的现象叫做生物富集。生物富集常用生物富集因子[1](bioconcentration factor,简称BCF)来表达。生物富集因子是化学品在水生生物和水体之间的平衡分配过程,其在生物体内(平衡)浓度与其水体中(平衡)浓度的比值称为生物富集因子,它反映水生生物对水体中有机物的吸收储存能力,是评价有机污染物生物累积性的重要指标。无论是确定持久性有机污染物(POPs),还是确定持久生物累积性有毒(PBT)污染物的清单,BCF都是一个不可或缺的参数。
BCF具有标准的测试方法[2],但实验测定BCF成本高、周期长,以及人力、经费、时间的限制,不可能对进入环境中的每种化学品都进行生物富集实验测定。因此,对化学品的生物富集因子进行计算机预测研究便显得尤为重要。迄今只有有限的有机化合物具有实测的BCF值。在环境科学技术中,定量结构-活性关系(QSAR)[3]指关联有机污染物的分子结构与其理化性质、环境行为和毒理学参数(统称为活性)的定量预测模型。QSAR可以弥补基础数据的缺失,降低昂贵的测试费用,减少动物实验。
在生态的角度上BCF的实验数据是很重要的,从监管的角度上来说也是很重要的。欧盟法规《化学品的注册、评估、授权和限制》(Registration, Evaluation, Authorization and Restriction of Chemicals, REACH)[4]要求每个化合物都要有与之对应的BCF值,从而推动了有机化合物BCF预测模型的建立。目前文献中已经报道了很多BCF预测模型,从最简单的log BCF与log KOW的线性模型[5-7]开始,有基于2D分子描述符的QSAR模型[8]、基于基线的BCF模型[9]、基于代谢的动力学模型[10]、基于拓扑指数的模型[11]、基于分子电性距离矢量(Molecular Electronegativity Distance Vector, MEDV)的模型[12]以及混合模型[13-14](几组模型的组合)等。同时也有很多预测模型整合到商业或者免费的软件中,例如ACD Labs[15]、VEGA[16]、OECD QSAR ToolBox[17]以及EPI suit[18]等。
本研究首先收集具有实验测定BCF值的化合物,然后使用7种不同的分子指纹结合5种机器学习方法来构建化合物BCF的定性分类预测模型,并使用多种方法来识别导致化合物发生生物富集的关键子结构作为预警。本研究所用方法也可用于环境毒理学中的其它毒性端点预测评价。
1 材料与方法(Materials and methods)
1.1 数据集准备
以美国环保局Estimation Program Interface (EPI) Suite[18]的BCFBAF程序中的Non-Ionic Training set[19]作为训练集,validation set作为外部验证集。其中训练集中有466个化合物,外部验证集中有158个化合物。依照生物富集的分类标准[20],将化合物分为低生物富集、中度生物富集以及高度生物富集。数据集的统计结果如表1所示。由于高生物富集化合物较少,因此在建模时将高生物富集和中度生物富集归为一类“易于生物富集”,将低生物富集归为一类“不易生物富集”,构建二分类定性预测模型。
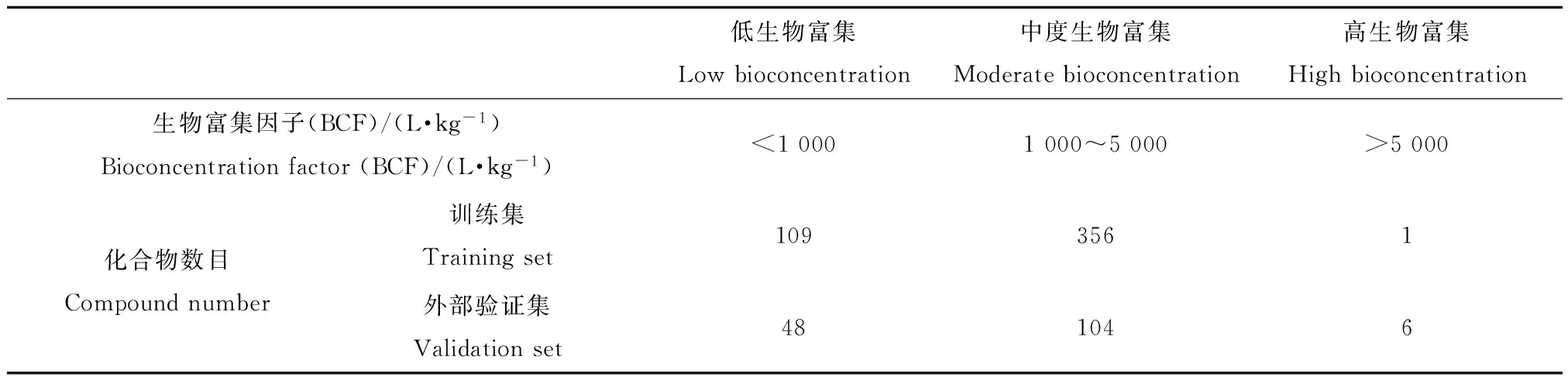
表1 数据集分布Table 1 Data sets and chemical toxicity categories
1.2 分子指纹计算
本研究使用7种分子指纹来表达化合物的分子结构,其中6种分子指纹分别是Fingerprint (FP,1024位)、EState fingerprint (EStFP,79位)、Extende fingerprint (ExtFP,1024位)、MACCS keys (MACCS,166位)、PubChem fingerprint (PubFP,881位)和Substructure fingerprint (FP4,307位),使用PaDEL-Descriptor[21]软件计算得到。第7种分子指纹使用ChemoTyper (CT,729位)软件[22]计算得到。在计算分子指纹之前数据集中所有化合物的SMILES先通过ChemAxon Standardizer[23]处理,其中的设置参数如下[24]:add explicit hydrogens, aromatize, clean 2D, remove fragment。
1.3 建模方法
本研究使用5种机器学习方法构建二分类模型。这5种机器学习方法分别是支持向量机(Support Vector Machine, SVM),最近邻居法(k-Nearest Neighbors, k-NN),朴素贝叶斯分类器(Naïve Bayes, NB),随机森林(Random Forest, RT),决策树(Decision Tree, C4.5)。这些方法均在Orange[25]软件中实现,Orange是一个基于Python脚本的数据挖掘和机器学习软件套装。为了测试模型的有效性和准确性,首先对训练集采用10倍交叉验证,检验模型的鲁棒性;然后进行外部验证集验证,检验模型的预测准确性。
支持向量机(SVM):本算法[26]是1995年由Vapnik和Cortes提出的一种统计学习算法。支持向量机通过对输入样本空间进行非线性映射转换,将输入空间变换为一种高维空间,从而利用线性分类平面来描述非线性的分类边界。这种非线性变换是通过核函数(Kernel Functions)实现的,在本研究中我们选取的核函数是RBF(Radial Basis Function)核函数,RBF核函数的优点包括可以将样本非线性的映射到高维空间,从而处理非线性问题,另外其只含有一个参数,形式简单。在Orange软件中,c值设定为10,g值设定为0.00212,且不勾选“Normalize data”选项。
最近邻居法(k-NN):又称k-近邻法,是著名的模式识别和统计学习方法之一[27],被广泛应用于文本分类、模式识别、图形图像以及空间分布等领域。该方法主要根据特征空间(描述空间)中最接近的样本进行分类,其基本思想是首先在多维向量中寻找与待分类样本最接近的k个邻居,然后根据这k个临近点的类别决定待分类样本所属的种类。本研究中选取的k值为9,距离公式选择欧几里得距离,同时选取了距离权重这个选项。
朴素贝叶斯分类器(NB):本方法[28]同样在化学分类模型中有着广泛的应用。它是贝叶斯分类器中简单而有效的一种,通过计算样本属于不同种类的概率,具有最大概率的类便是该样本所属的类。朴素贝叶斯分类器的优点是分类过程中占用的计算资源很少,分类结果很稳定,鲁棒性很好。在Orange软件中使用默认参数设置。
决策树(C4.5):本方法在众多的模式识别方法中是最经典和最古老的方法之一。C4.5是一种以实例为基础的归纳学习算法,从无序的训练样本中,归纳出分类的标准,其基本构成包括决策结点,分支和叶结点。决策树的目标是根据简单的几个变量(描述符)输入建立一种简单的规则预测一个目标值。朴素贝叶斯算法在Orange软件中使用的是默认参数设置。
随机森林(RF):本方法是一个包含多个决策树的分类器,并且其输出的类别是由个别树输出的类别的众数而定。在Orange软件中,number of trees in forest参数设置为100。
1.4 预测模型评价
所有的模型均采用10倍交叉验证(10-fold cross validation)和外部验证集验证。通过计算如下变量的化合物数目:真阳性(True Positives, TP)、真阴性(True Negatives, TN)、假阳性(False Positives, FP)和假阴性(False Negatives, FN)。然后计算每个模型的敏感性、特异性和预测准确率。敏感性(Sensitivity, SE)表示的是对中/高生物富集化合物预测正确的百分比;特异性(Specificity, SP)表示的是对低生物富集化合物预测正确的百分比;整体预测正确率(Accuracy, Q)表示的是中/高生物富集化合物和低生物富集化合物都预测正确的百分比。SE、SP和Q值均在0到1之间,值越高,表明模型的预测能力越好。马修斯相关系数(MCC)代表模型的整体预测能力,MCC的范围处于-1与+1之间。当某个预测模型的MCC值等于+1时,这就代表着这个模型的预测能力达到了最优水平;相反,如果某个预测模型的MCC值等于-1,那么它得到的则是最差的预测结果。具体的计算方程如下[29]:
另外,受试工作特征曲线(ROC曲线)也被用来表征模型的预测能力[30]。ROC曲线能形象地呈现出模型的预测能力,一般曲线越往上偏,表明模型预测的精准度越高。工作特性曲线下面积(AUC)值越大,表示模型预测能力越高。最理想的模型AUC=1,随机产生的模型AUC=0.5。
1.5 警示子结构识别
本研究使用信息增益(Information gain,简称IG)[31]和子结构片段频率分析[32]来获得警示子结构。如果一个子结构在高/中生物富集化合物中出现的频率比低生物富集化合物中出现的频率更高,这个子结构就可以被认为是易于产生生物富集的特征子结构,需要警惕[33]。警示子结构是由毒性机理总结而来,是一种很重要的预测工具[34]。生物富集化合物中,片段出现的频率定义为如下公式计算:

其中,N子结构类别表示的是在中/高生物富集化合物中包含有此片段的总数目,N总数表示数据集中所有化合物数目,N子结构总数是表示包含该子结构片段的所有化合物数目,N类别是表示在中/高生物富集化合物的数目。
我们也使用了免费软件KNIME (Konstanz Information Miner)[35]中的MoSS模块和ChemoTyper软件搜寻一系列分子数据集中频繁出现的分子结构片段。KNIME的MoSS模块中,“minimum fragment size”是一个重要的参数,经过测试比较,我们把这个值设定为8;“maximum fragment size”设定为100,“minimum focus support in %”设定为8,“maximum complement support in %”设定为20,其他参数使用默认设置。ChemoTyper软件可以根据化合物的结构信息搜索警示子结构。
2 结果(Results)
2.1 数据集分析
影响模型质量好坏的关键因素是数据集的质量,本研究中使用的是BCFBAF软件中的数据集。通过化合物的分子量(molecule weight)和ALogP来定义数据集(包含训练集以及外部验证集)学空间分布,如图1所示。从图中可以看出,外部验证集化合物的化学空间与训练集分子的化学空间类似,分布在同一区域内。为了进一步研究数据集的应用域,我们另使用5种物理描述符(AlogP、分子质量、溶解度、氢键受体数目和氢键供体数目)雷达图[36]来定义(图2)。如图2中所示,分子质量的最小值为68.074,最大值为959.168;AlogP的最小值为-1.661,最大值为到10.874;溶解度的最小值为-15.489,最大值为1.094;氢键受体数目为0到9;氢键供体数目为0到3,这些数据说明我们的数据集具有较大的应用域。
2.2 模型构建
本研究使用7种分子指纹结合5种机器学习方法构建了35个二分类模型。通过分析训练集10倍交叉验证的结果(表2)发现,5种机器学习方法在预测生物富集方面存在一定的差异性,使用不同的分子指纹的建模结果也略有差异。其中使用SVM和k-NN这两种机器学习方法时,模型的鲁棒性较好。例如,在MACCS-kNN和MACCS-SVM模型中,Q值分别为0.856和0.850,AUC值分别为0.869和0.884,MCC值分别为0.586和0.546,均高于使用其他机器学习方法建立模型的结果。
2.3 外部验证集验证
通过分析训练集的10倍交叉验证结果发现使用SVM和k-NN机器学习方法时模型的鲁棒性较好,因此对这两种机器学习方法构建的模型进行外部验证集验证(见表3)。在这7种分子指纹中,使用CT、ExtFP、PubFP和MACCS这四种分子指纹描述分子特征时模型的预测结果较好,即模型CT-SVM、ExtFP-SVM和PubFP-SVM具有最好的预测准确度。在这四个模型中,Q值分别为0.854、0.842、0.835和0.810,SE值分别为0.9、0.918、0.918和0.882,SP值分别为0.75、0.667、0.646和0.646;AUC值分别为0.910、0.910、0.911和0.890(图3),MCC值分别为0.654、0.614、0.597和0.541。通过对比分析Q、SE、SP、AUC以及MCC值,发现CT-SVM模型为最优模型。
2.4 警示子结构识别
通过信息增益分析,我们找到了10个潜在的具有致生物富集的警示子结构,分别是芳基氯化物(arylchloride)、二芳基醚(diarylether)、芳基溴化物(arylbromide)、卤素缩醛类似物(halogen acetal like)、氯代烷烃(chloroalkene)、稠环(annelated rings)、羧酸酯(carboxylic ester)、叔碳(tertiary carbon)、桥环(bridged rings)、仲碳(secondary carbon)。这些子结构的命名均基于FP4命名规则,具体结构及其信息增益值见表4。
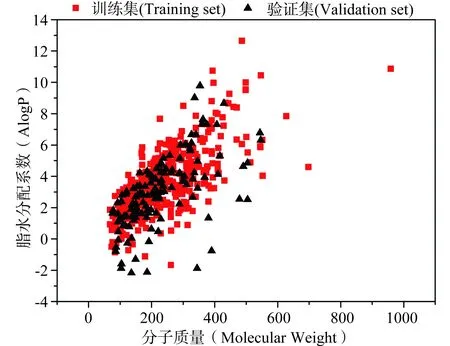
图1 训练集化合物和外部验证集化合物的空间分布Fig. 1 Diversity analysis of chemicals in the training set and validation set

图2 5个物理描述符的雷达分析图 Fig. 2 The radar chart of five physicochemical descriptors (AlogP, Molecular Weight, Solubility, H-Acceptors and H-Donors)
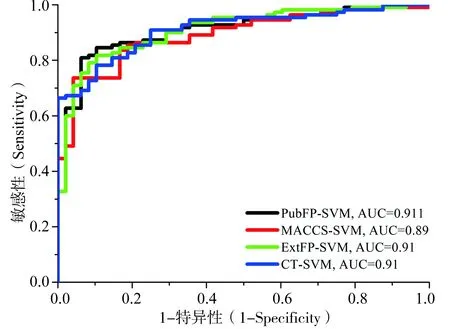
图3 模型CT-SVM、ExtFP-SVM、MACCS-SVM和 PubFP-SVM的受试者工作特性曲线图Fig. 3 Representation of receiver operating characteristics (ROC) curve for the validation set in model CT-SVM, ExtFP-SVM, MACCS-SVM and PubFP-SVM
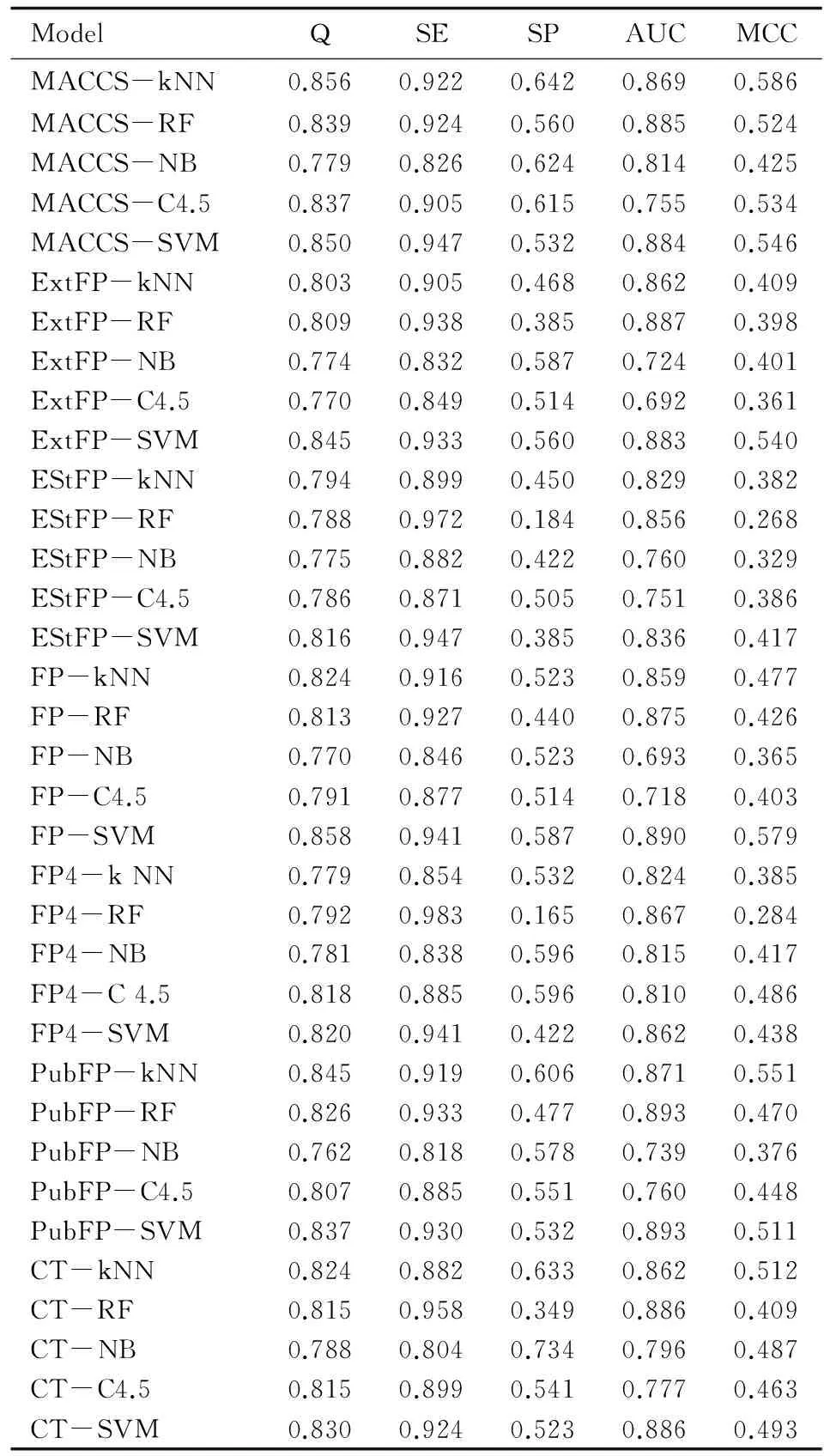
表2 训练集的10倍交叉验证结果Table 2 The performance of 10-fold cross validation in training set
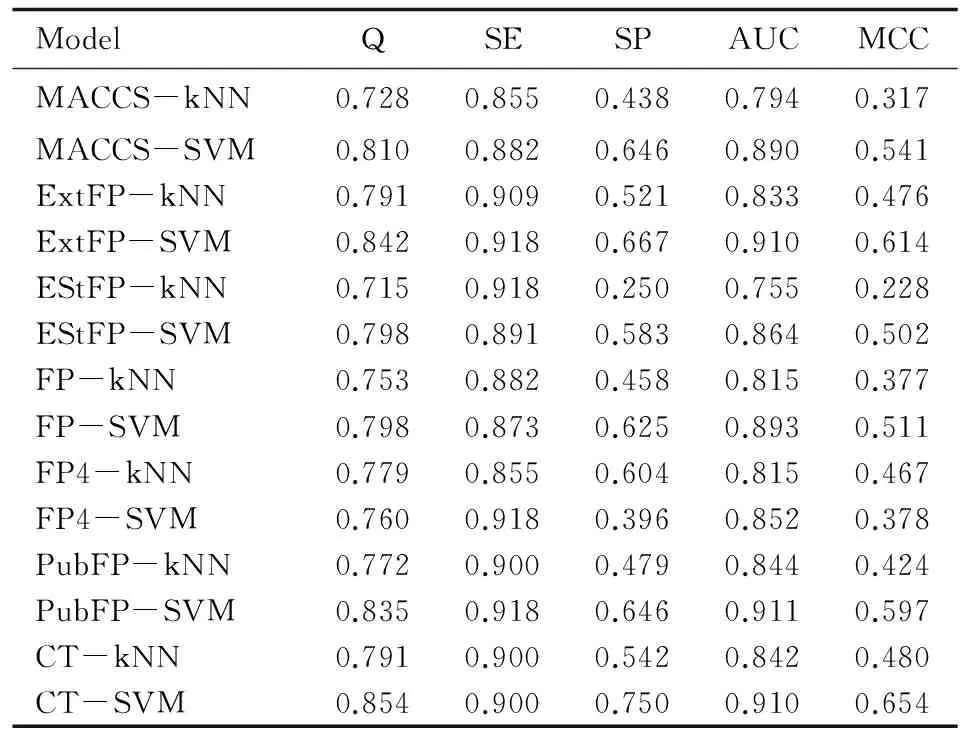
表3 SVM和k-NN构建模型的外部验证集结果Table 3 The performance of models using SVM and k-NN methods for validation set
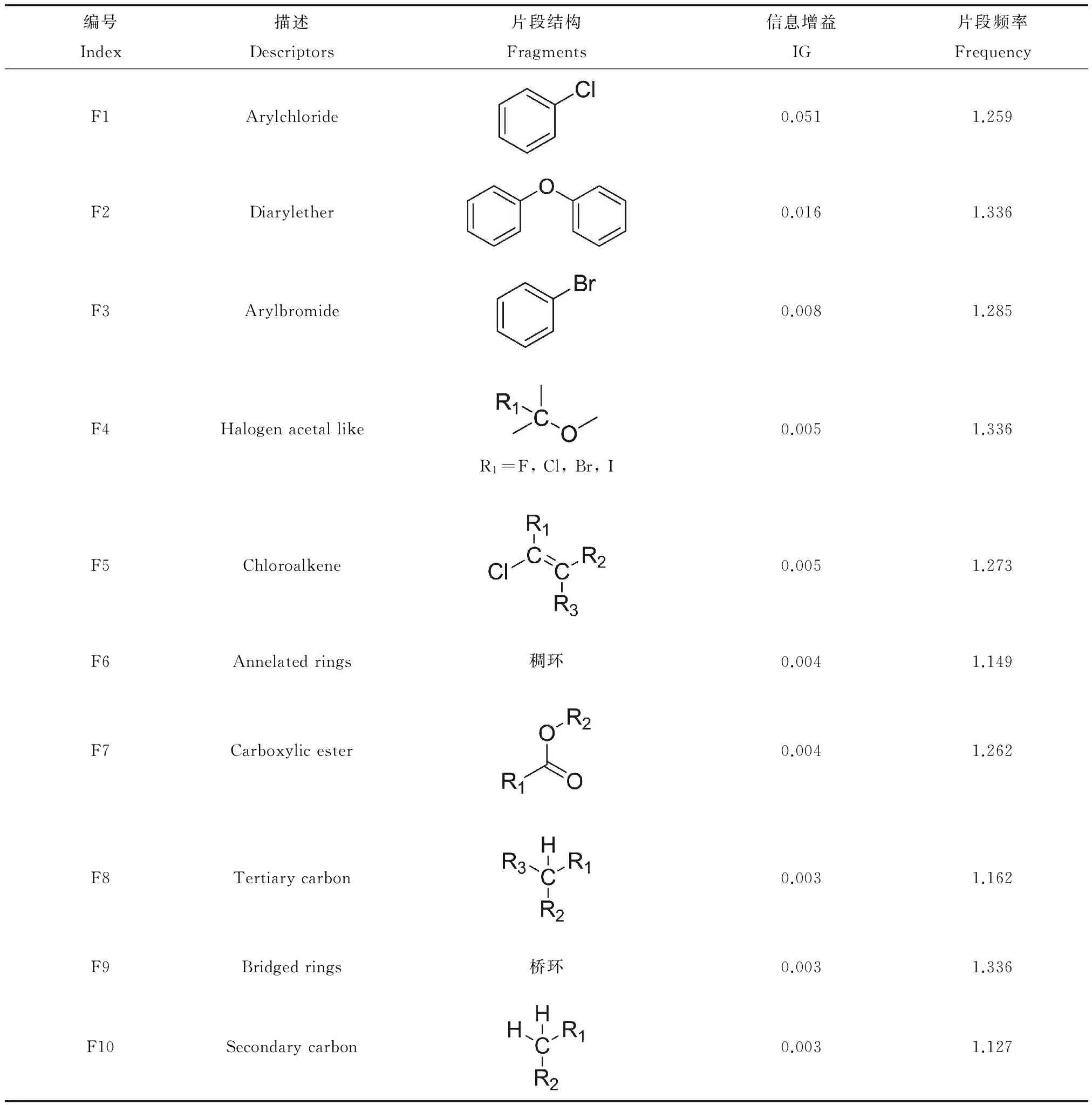
表4 警示子结构的频率分析和信息增益结果Table 4 The common substructure alerts identified using IG methods and frequency analysis
通过KNIME软件的MoSS模块检索生物富集化合物结构,得到了10个子结构,分别为1-氯-3-甲基苯、1,3-二氯苯、异丙苯、1,2,4-三氯苯、1,2,3-三氯苯、1,2-二氯苯、硝基苯、苯甲醚、乙苯和1,4-二氯苯。上述子结构及包含对应结构的代表性化合物分子见表5。从表5中可以看出,这10个子结构在生物富集化合物中出现的频率最大,同样给了我们警示的作用。
使用ChemoTyper软件根据数据集的结构信息搜寻数据集中的警示子结构,找到10个警示子结构,见表6。表6中“X”和“?”均表示卤素原子,当“X”和“?”为氯原子时,这10个警示子结构为1,4-二氯苯、1,2,4-三氯苯、1,2,3-三氯苯、3-氯酚、二苯基甲烷、1,3,5-三氯苯、1-氯-2-(3-氯苯氧基)苯、1,2,3-三氯、1,2-二氯乙烯和2-氯-2-甲基丙烷。这些子结构中,有7个为芳香族结构片段,且在苯环上均连接有卤素原子;3个为烷烃类结构,均为卤代烷烃。
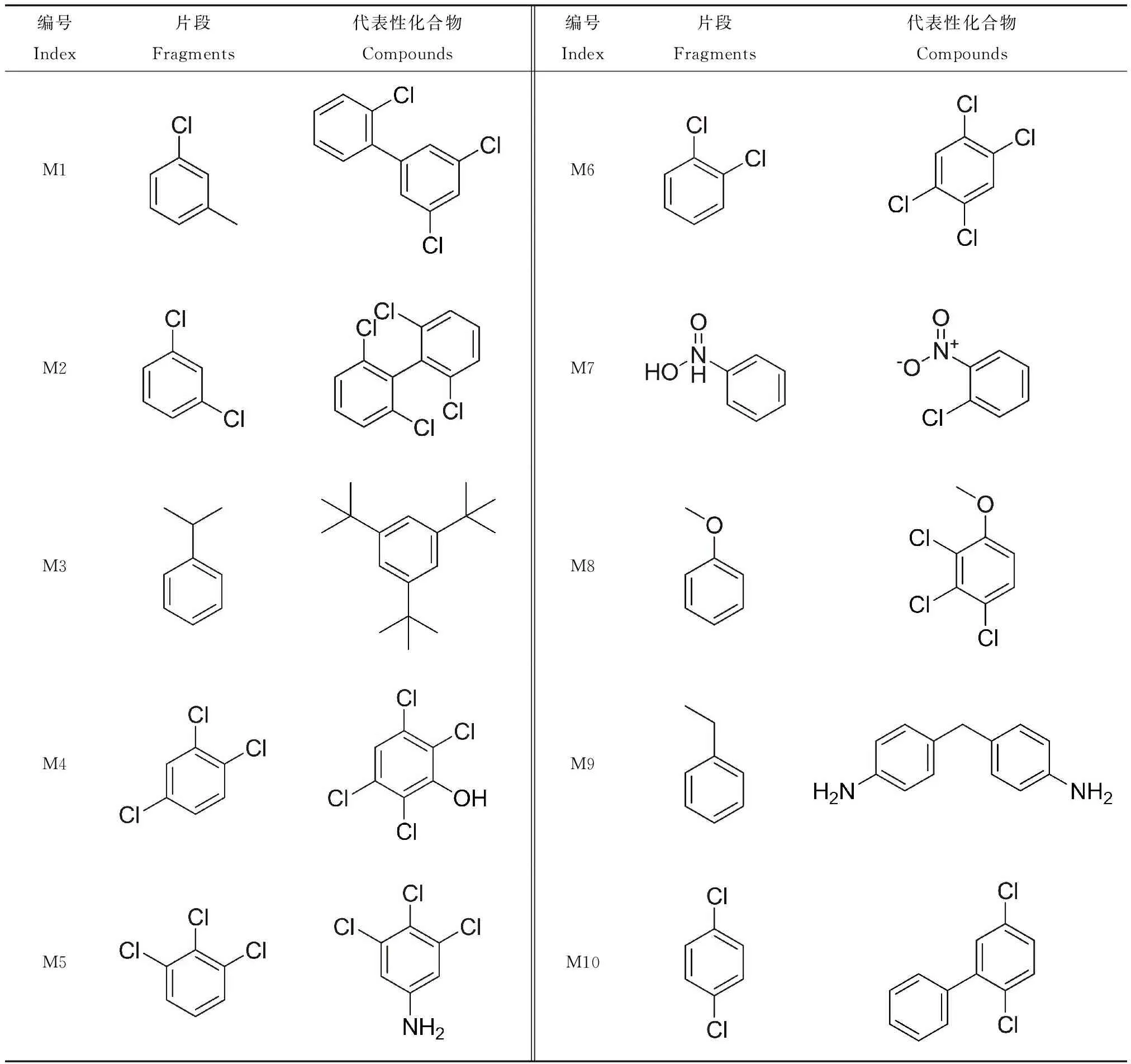
表5 MoSS分析结果以及包含其警示结构的代表性化合物Table 5 The results of MoSS searched for structural alerts and representative structures
3 讨论(Discussion)
3.1 模型结果分析
在我们的研究中,使用了5种不同的机器学习方法(SVM、C4.5、RF、k-NN和NB)。通过分析表2中的10倍交叉验证结果(Q、SE、SP、AUC以及MCC值),可以看出整体趋势上,在使用同一分子指纹描述分子特性时,SVM和k-NN两种机器学习方法显示了良好的预测精度。MCC值代表模型的整体预测能力,在使用SVM和k-NN建模时,模型的MCC值要明显高于其他机器学习方法。例如模型CT-kNN和CT-SVM的MCC值分别为0.512和0.493,明显优于其他机器学习方法。
SVM具有很强的拟合非线性关系的能力,并在一定程度上成为预测准确度的“黄金标准”。k-NN算法之所以在10倍交叉验证预测准确率较高是由其算法的特殊性和生物富集这个特殊的毒性端点所决定的。一个化合物被预测为易于富集化合物还是不易富集化合物,主要根据它附近的邻居化合物的富集与否所决定。被分为相同类的化合物之间结构具有相似性。由于这个因素,如果数据库包含的化合物数量足够大和化合物结构足够多样,那么以k-NN作为建模方法建立的模型去预测化合物的生物富集因子,就能够获得很高的预测准确度。
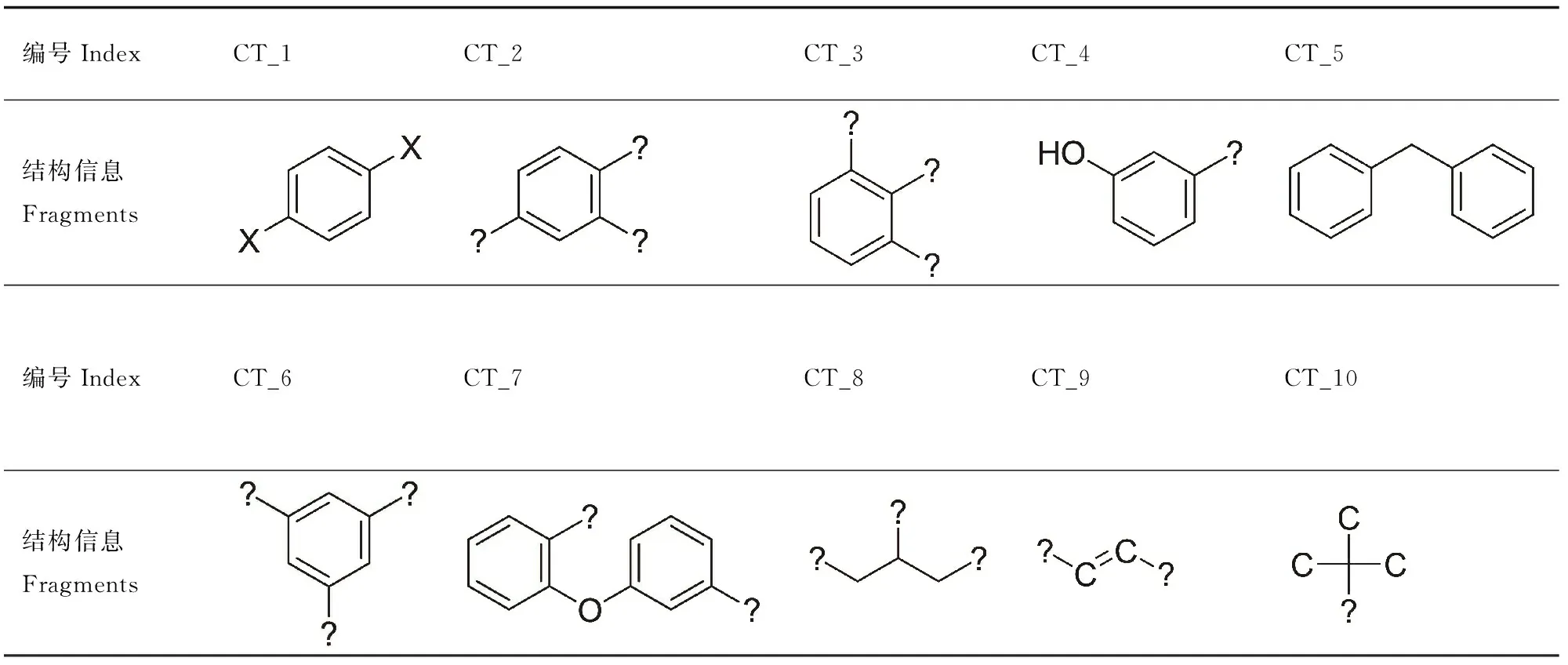
表6 ChemoTyper分析结果Table 6 The results of ChemoTyper analysis searched for structural alerts
注:“X”和“?”均表示卤素。
Note: “X” and “?” means halogen.
当使用相同的建模方法,不同的分子指纹作为属性变量的建模结果略有差异。如表2中所示,当使用k-NN建模时,模型MACCS-kNN、ExtFP-kNN、EStFP-kNN、FP-kNN、FP4-kNN、PubFP-kNN和CT-kNN的Q值分别为0.856、0.803、0.794、0.824、0.779、0.845和0.824。使用MACCS、ExtFP、FP、PubFP和CT这五个分子指纹的建模结果要优于EStFP、FP4。EstFP分子指纹的长度为79,在这79个分子片段中仅有35个片段用来描述模型的结构信息;FP4分子指纹中含有307个片段,但用来描述模型结构信息的片段仅有93位。大量的信息丢失是导致预测准确度低的重要因素,可能是其不具备优秀的特征来表征多样数据集中的分子结构特性。
3.2 与EPI中的预测软件进行比较
BCFBAF是由EPA研究开发预测BCF的一款软件,现已整合到EPI Suite中。本研究建模使用的数据为BCFBAF软件中的non-ionic training set,外部验证集也为BCFBAF软件中的validation set。BCFBAF软件对外部验证集的预测结果为Q值为0.854、SE值为0.9以及SP值为0.75。通过与BCFBAF软件的预测准确度的比较显示,我们构建的模型CT-SVM与BCFBAF的预测准确度与其相当,从而进一步证明了本研究所用建模方法的可行性。
3.3 警示子结构分析
影响生物富集的因素有很多,例如生物物种特性、化合物的性质、化合物的浓度和作用时间,以及环境因素等都是影响生物富集的主要因素。本研究采用信息增益方法、KNIME中的MoSS模块以及ChemoTyper软件找到了一系列警示子结构,期望从化合物结构上寻找易于引起生物富集的原因。化合物的稳定性和脂溶性是引起生物富集的重要条件。例如DDT化学稳定性强,为脂溶性物质,易被吸收和积累在脂肪中。类似的化合物有机氯农药、多氯联苯、甲基汞等化合物。我们使用信息增益的方法找到的10个警示子结构中,有3个含有卤素原子(见表4),分别为芳基氯、芳基溴和氯代烷烃;使用KNIME中的MoSS模块找到的10个警示子结构中,有6个片段含有卤素原子(见表5);使用ChemoTyper软件找到的10个警示子结构中,9个片段含有卤素原子(见表6)。以上警示子结构均和文献报道的已知生物富集化合物的结构特征相吻合,证明了我们方法的可靠性。
从结果多样性上分析以上三种方法找到的警示结构发现,使用KNIME中的MoSS模块找到的10个警示子结构均为芳香族化合物,使用ChemoTyper软件找到的10个警示子结构中有7个子结构为芳香族化合物,而使用信息增益的方法找到的10个警示子结构属于不同类的化合物。因此使用信息增益的方法寻找到的警示子结构从结构多样化的角度,优于KNIME中的MoSS模块和ChemoTyper软件。
本研究中我们使用7种不同的分子指纹结合5种机器学习方法构建了具有高预测准确度的二分类生物富集因子预测模型,使用10倍交叉验证的方法验证模型的鲁棒性。其中四个模型(CT-SVM、ExtFP-SVM、PubChem-SVM和MACCS-SVM)对中/高生物富集和低生物富集化合物都具有很高的预测准确度,从而保证了模型具有一定的实用性。另外,与BCFBAF软件的预测结果相对比,发现我们构建的模型CT-SVM与BCFBAF软件的预测结果相当,从而进一步证明了使用分子指纹描述分子特征构建分类模型方法的可行性。在本研究的最后,采用信息增益子结构碎片分析、KNIME软件的MoSS模块分析和ChemoTyper软件分析了生物富集的特权子结构碎片和警示结构,对生态系统安全评估具有一定的指导意义。生物富集过程并非一个简单、机械的分配过程,它受到很多因素的制约和影响,例如生物物种的特性、污染物的性质、污染物的浓度及其作用时间,以及环境等都是影响生物富集的因素。因此只有应用多参数分析的方法,在大量的实验数据的基础上,才能寻找出更为合理的估算方法。
[1] Arnot J A, Gobas F A. A review of bioconcentration factor (BCF) and bioaccumulation factor (BAF) assessments for organic chemicals in aquatic organisms [J]. Environmental Reviews, 2006, 14(4): 257-297
[2] OECD. OECD Guidelines for Testing of Chemicals. TG 305: Bioaccumulation in Fish: Aqueous and Dietary Exposure [R]. OECD, 2012
[3] Cherkasov A, Muratov E N, Fourches D, et al. QSAR modeling: Where have you been? Where are you going to? [J]. Journal of Medical Chemistry, 2014, 57(12): 4977-5010
[4] European Chemicals Agency. Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC [S]. European Chemicals Agency, 2007
[5] Weisbrod A V, Burkhard L P, Arnot J, et al. Workgroup report: Review of fish bioaccumulation databases used to identify persistent, bioaccumulative, toxic substances [J]. Environmental Health Perspectives, 2007, 115(2): 255-261
[6] Saçan M T, Erdem S S, Ozpinar G A, et al. QSPR study on the bioconcentration factors of nonionic organic compounds in fish by characteristic root index and semiempirical molecular descriptors [J]. Journal of Chemical Information and Modeling, 2004, 44(3): 985-992
[7] Neely W B, Branson D R, Blau G E. Partition coefficient to measure bioconcentration potential of organic chemicals in fish [J]. Environmental Science & Technology, 1974, 8(13): 1113-1115
[8] Roy K, Sanyal I, Roy P P. QSPR of the bioconcentration factors of non-ionic organic compounds in fish using extended topochemical atom (ETA) indices [J]. SAR and QSAR in Environmental Research, 2006, 17(6): 563-582
[9] Dimitrov S, Dimitrova N, Parkerton T, et al. Base-line model for identifying the bioaccumulation potential of chemicals [J]. SAR and QSAR in Environmental Research, 2005, 16(6): 531-554
[10] Stadnicka J, Schirmer K, Ashauer R. Predicting concentrations of organic chemicals in fish by using toxicokinetic models [J]. Environmental Science & Technology, 2012, 46(6): 3273-3280
[11] Khadikar P V, Singh S, Mandloi D, et al. QSAR study on bioconcentration factor (BCF) of polyhalogented biphenyls using the PI index [J]. Bioorganic & Medicinal Chemistry, 2003, 11(23): 5045-5050
[12] Cui S H, Yang J, Liu S S, et al. Predicting bioconcentration factor values of organic pollutants based on MEDV descriptors derived QSARs [J]. Science in China Series B: Chemistry, 2007, 50(5): 587-592
[13] Zhao C, Boriani E, Chana A, et al. A new hybrid system of QSAR models for predicting bioconcentration factors (BCF) [J]. Chemosphere, 2008, 73(11): 1701-1707
[14] Gissi A, Nicolotti O, Carotti A, et al. Integration of QSAR models for bioconcentration suitable for REACH [J]. The Science of the Total Environment, 2013, 456-457: 325-332
[15] ACD Labs. ACD Labs homepage [OL]. [2014-12-04]. http://www.acdlabs.com/home/
[16] VEGA. VEG homepage [OL]. [2014-12-04]. http://www.vega-qsar.eu/
[17] OECD. QSAR ToolBox [OL]. [2014-12-04]. http://www.qsartoolbox.org/
[18] Estimation Program Interface (EPI) Suite [OL]. [2014-12-04]. http://www.epa.gov/opptintr/exposure/pubs/episuite.htm
[19] US Environmental Protection Agency. EPI Suite Data [OL]. [2014-12-04]. http://esc.syrres.com/interkow/EpiSuiteData.htm
[20] Costanza J, Lynch D G, Boethling R S, et al. Use of the bioaccumulation factor to screen chemicals for bioaccumulation potential [J]. Environmental Toxicology and Chemistry, 2012, 31(10): 2261-2268
[21] Yap C W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints [J]. Journal of Computational Chemistry, 2011, 32(7): 1466-1474
[22] ChemoTyper Community. ChemoTyper website [OL]. [2014-12-04]. https://chemotyper.org/
[23] ChemAxon. ChemAxon website [OL]. [2014-12-04]. http://www.chemaxon.com
[24] Fourches D, Muratov E, Tropsha A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research [J]. Journal of Chemical Information and Modeling, 2010, 50: 1189-1204
[25] Orange website. Orange website [OL]. [2014-12-04]. http://orange.biolab.si/
[26] Cortes C, Vapnik V. Support-Vector Networks [J]. Machine Learning, 1995, 20(3): 273-379
[27] Itskowitz P, Tropsha A. kappa Nearest neighbors QSAR modeling as a variational problem: Theory and applications [J]. Journal of Chemical Information and Modeling, 2005, 45(3): 777-785
[28] Watson P. Naïve Bayes classification using 2D pharmacophore feature triplet vectors [J]. Journal of Chemical Information and Modeling, 2008, 48(1): 166-178
[29] Cheng F X, Yu Y, Zhou Y D, et al. Insights into molecular basis of cytochrome p450 inhibitory promiscuity of compounds [J]. Journal of Chemical Information and Modeling, 2011, 51(10): 2482-2495
[30] Baldi P, Brunak S, Chauvin Y, et al. Assessing the accuracy of prediction algorithms for classification: An overview [J]. Bioinformatics, 2000, 16(5): 412-424
[31] Shen J, Cheng F X, Xu Y, et al. Estimation of ADME properties with substructure pattern recognition [J]. Journal of Chemical Information and Modeling, 2010, 50(6): 1034-1041
[32] Jensen B F, Vind C, Padkjaer S B, et al. In silico prediction of cytochrome P450 2D6 and 3A4 inhibition using Gaussian kernel weighted k-nearest neighbor and extended connectivity fingerprints, including structural fragment analysis of inhibitors versus noninhibitors [J]. Journal of Medicinal Chemistry, 2007, 50(3): 501-511
[33] Kruhlak N L, Contrera J F, Benz R D, et al. Progress in QSAR toxicity screening of pharmaceutical impurities and other FDA regulated products [J]. Advanced Drug Delivery Reviews, 2007, 59(1): 43-55
[34] Benigni R, Bossa C. Structure alerts for carcinogenicity, and the Salmonella assay system: A novel insight through the chemical relational databases technology [J]. Mutation Research, 2008, 659(3): 248-261
[35] KNIME. KNIME website [OL]. [2014-12-04]. http://www.knime.org/
[36] Cheng F X, Shen J, Xu Y, et al. In silico prediction of Tetrahymena pyriformis toxicity for diverse industrial chemicals with substructure pattern recognition and machine learning methods [J]. Chemosphere, 2011, 82(11): 1636-1643
◆
InSilicoPrediction of Chemical Bioconcentration Factor
Sun Lu, Chen Yingjie, Wu Zengrui, Li Weihua, Liu Guixia, Philip W. Lee, Tang Yun*
Shanghai Key Laboratory of New Drug Design, School of Pharmacy, East China University of Science and Technology, Shanghai 200237, China
5 December 2014 accepted 9 January 2015
Bioconcentration is an important endpoint in evaluation of chemical adverse effects on ecosystems. In this study, in silico methods were used to predict chemical bioconcentration factor (BCF). At first a data set containing 624 chemicals with BCF values was collected from the Estimation Program Interface Suite of the U. S. Environmental Protection Agency. Using seven fingerprints to represent the molecules, binary classification models were developed with five machine learning methods, including support vector machine (SVM), C4.5 decision tree (C4.5 DT), k-nearest neighbors (kNN), random forest (RF), and Naïve Bayes (NB). Reliable predictive models were then obtained and validated by 10-fold cross validation and external validation set. Among them, the model built by SVM with ChemoTyper fingerprint performed best, with predictive accuracy up to 85.4%. Moreover, some substructures were identified to be key for bioconcentration via several methods, such as arylchloride, diarylether, chloroalkene, and so on. The approaches used in this study provide a useful tool for environmental risk assessment of chemicals.
bioconcentration factor; in silico prediction; binary classification models; substructural alerts; environmental toxicology
国家自然科学基金(No. 81373329);学科创新引智计划即111计划(No. B07023)
孙露(1989-),女,硕士,研究方向为计算机辅助药物设计、药物信息学和计算毒理学,E-mail: sunlu900326@yeah.net;
*通讯作者(Corresponding author), E-mail: ytang234@ecust.edu.cn
10.7524/AJE.1673-5897.20141205001
2014-12-05 录用日期:2015-01-09
1673-5897(2015)2-173-10
X171.5
A
唐赟(1968-),男,博士,教授,主要研究方向为计算机辅助药物设计、药物信息学、计算生物学和计算毒理学,已发表学术论文100余篇。
孙露, 陈英杰, 吴曾睿, 等. 有机化合物生物富集因子的计算机预测研究[J]. 生态毒理学报, 2015, 10(2): 173-182
Sun L, Chen Y J, Wu Z R, et al. In silico prediction of chemical bioconcentration factor [J]. Asian Journal of Ecotoxicology, 2015, 10(2): 173-182 (in Chinese)
猜你喜欢
中学生数理化·中考版(2021年12期)2021-12-31
中学生数理化·中考版(2021年11期)2021-12-06
科技信息·学术版(2021年18期)2021-10-25
小哥白尼(趣味科学)(2021年11期)2021-02-28
小天使·一年级语数英综合(2020年10期)2020-12-16
中学化学(2017年6期)2017-10-16
西安建筑科技大学学报(自然科学版)(2016年1期)2016-11-08
自动化学报(2016年8期)2016-04-16
小型内燃机与车辆技术(2015年4期)2015-10-22
青少年科技博览(中学版)(2015年7期)2015-08-12
