
HPLC法测定黄鱼中酸性橙Ⅱ染料
2015-06-01 10:24:37冯晓青刘华良阮丽萍杭学宇
分析仪器 2015年4期
冯晓青 刘华良 阮丽萍 王 芹 王 露 汪 怡 杭学宇
(1.淮安市疾病预防控制中心, 淮安 223001;2.江苏省疾病预防控制中心,南京 210009)
仪器应用
HPLC法测定黄鱼中酸性橙Ⅱ染料
冯晓青1刘华良2阮丽萍2王 芹1王 露1汪 怡1杭学宇1
(1.淮安市疾病预防控制中心, 淮安 223001;2.江苏省疾病预防控制中心,南京 210009)
建立了操作简便、灵敏度较高的HPLC测定黄鱼中酸性橙Ⅱ的方法。样品经均质,取2 g粉碎样品,乙醇震荡提取5 min,提取液经聚酰胺固相萃取柱分离净化,采用C18色谱柱(250 mm×4.6 mm×5 μm)分离,以甲醇-乙酸铵溶液梯度洗脱,流速为1 mL/min,设定柱温30 ℃,进样体积为10 μL,二极管阵列检测器在λ=485 nm处检测酸性橙Ⅱ(λ=485 nm)。酸性橙Ⅱ在0.1 μg/mL~20 μg/mL的范围内,呈良好的线性响应,线性方程为y=34.94x-2.697,r=0.9998。酸性橙Ⅱ检出限为0.08 μg/mL。样品添加浓度为0.5 mg/kg~15 mg/kg时,方法回收率为96%~99%,相对标准偏差为3.4%~7.3%。该法用于黄鱼中的酸性橙Ⅱ的含量测定,操作简便,定量准确可靠。
酸性橙Ⅱ 黄鱼 HPLC
色素酸性橙Ⅱ又名金橙Ⅱ,二号橙,桔黄橙,酸性金黄Ⅱ或酸性艳橙GR,俗名金黄粉,化学名为2-萘酚偶氮对苯磺酸钠。酸性橙Ⅱ是一种工业染料,主要用于蚕丝、羊毛织品和皮革的染色,禁止作为食品添加剂使用。在食品中违规添加酸性橙Ⅱ,极大危害消费者的身体健康。卫生部重新汇总发布的《食品中可能违法添加的非食用物质和易滥用的食品添加剂名单》中“产品类别”增加“鲍汁、腌卤肉制品、红壳瓜子、辣椒面和豆瓣酱”。但因酸性橙为化工原料,目前对食品中酸性橙Ⅱ的检测在国家标准及地方标准中仍是空白。
文献报道酸性橙Ⅱ的测定方法有纸层析法、分光光度法、反相高效液相色谱法和液相色谱串联质谱法等[1-8]。本实验旨在通过研究和优化酸性橙Ⅱ的试验参数,建立快速、准确、实用的高效液相色谱检测方法,为黄鱼的市场质量监督以及安全检测提供依据。
1 材料与方法
1.1 仪器与试剂
高效液相色谱仪(Agilent 1260,美国安捷伦公司),二极管阵列检测器及数据处理器;超声波清洗器(KH-250DB,昆山禾创公司);食品粉粹机(欧科电器有限公司);离心机(H-1850R,湘仪仪器厂);漩涡混匀器(SI-T256,美国Scientific Industries公司);氮吹仪(DB-3D,英国TECHENE公司);固相萃取装置(DG-12,美国Supelco公司)。酸性橙Ⅱ标准品(Sigma公司,批号:213031462);甲醇(色谱纯,批号:1205254);乙醇、氨水等均为分析纯;聚酰胺固相萃取柱(1000 mg/6 mL,北京康农兴牧科技发展中心);黄鱼购自市场。
1.2 实验方法
1.2.1 样品处理
将黄鱼样品打碎成肉泥状,混匀后准确称取2 g粉碎样品于15 mL具塞离心管中,加入10 mL乙醇震荡提取5 min,以5000 r/min的速度离心5min,取上清液于另一离心管中,在残渣中加乙醇10mL,再重复提取一次,合并两次上清液,备用。聚酰胺固相萃取柱依次用3mL水、3mL乙醇活化,提取上清液过柱,控制流速小于2mL/min。用3 mL水和3 mL 乙醇淋洗,乙醇-氨水-水溶液4 mL洗脱,收集洗脱液,于80 ℃氮气氛围下吹干,用1.0 mL流动相溶解残余物,涡旋1 min,以10000 r/min的速度离心5 min,取上清液,供高效液相色谱测定。
1.2.2 色谱条件
色谱柱:C18柱(250 mm×4.6 mm×5 μm)或性能相当者;流动相:甲醇-0.02 mol/L乙酸铵溶液;流速:1 mL/min,梯度洗脱见表1;柱温:30 ℃;检测波长:484 nm;进样体积:10 μL。
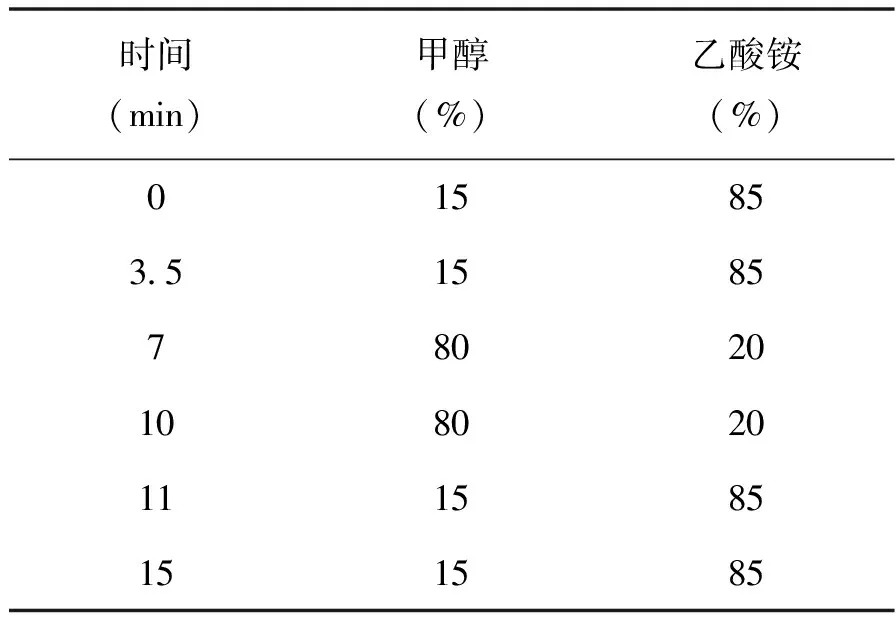
表1 流动相梯度洗脱条件
1.2.3 标准曲线绘制
精确称取0.100 g酸性橙Ⅱ,于100 mL容量瓶中,用甲醇溶解并稀释至刻度,配制成浓度为1 mg/mL的酸性橙Ⅱ标准贮备液。吸取标准贮备液,用流动相稀释配制成浓度分别为0.1 μg/mL、0.2 μg/mL、0.5 μg/mL、1.0 μg/mL、2.0 μg/mL、5.0 μg/mL、10.0 μg/mL、20.0 μg/mL的系列标准溶液,绘制工作曲线。
1.2.4 测定方法
取试样溶液和相应的对照溶液,作单点或多点校准,按外标法,以峰面积计算。对照溶液及试样溶液中酸性橙Ⅱ的响应值应在仪器检测的线性范围之内。
2 结果与讨论
2.1 波长选择
取5.0 μg/mL的酸性橙Ⅱ标准液,按1.2.2的色谱条件进样,在波长为200 nm~600 nm的范围内对酸性橙Ⅱ进行全扫描(图1),选定484 nm作为检测波长。
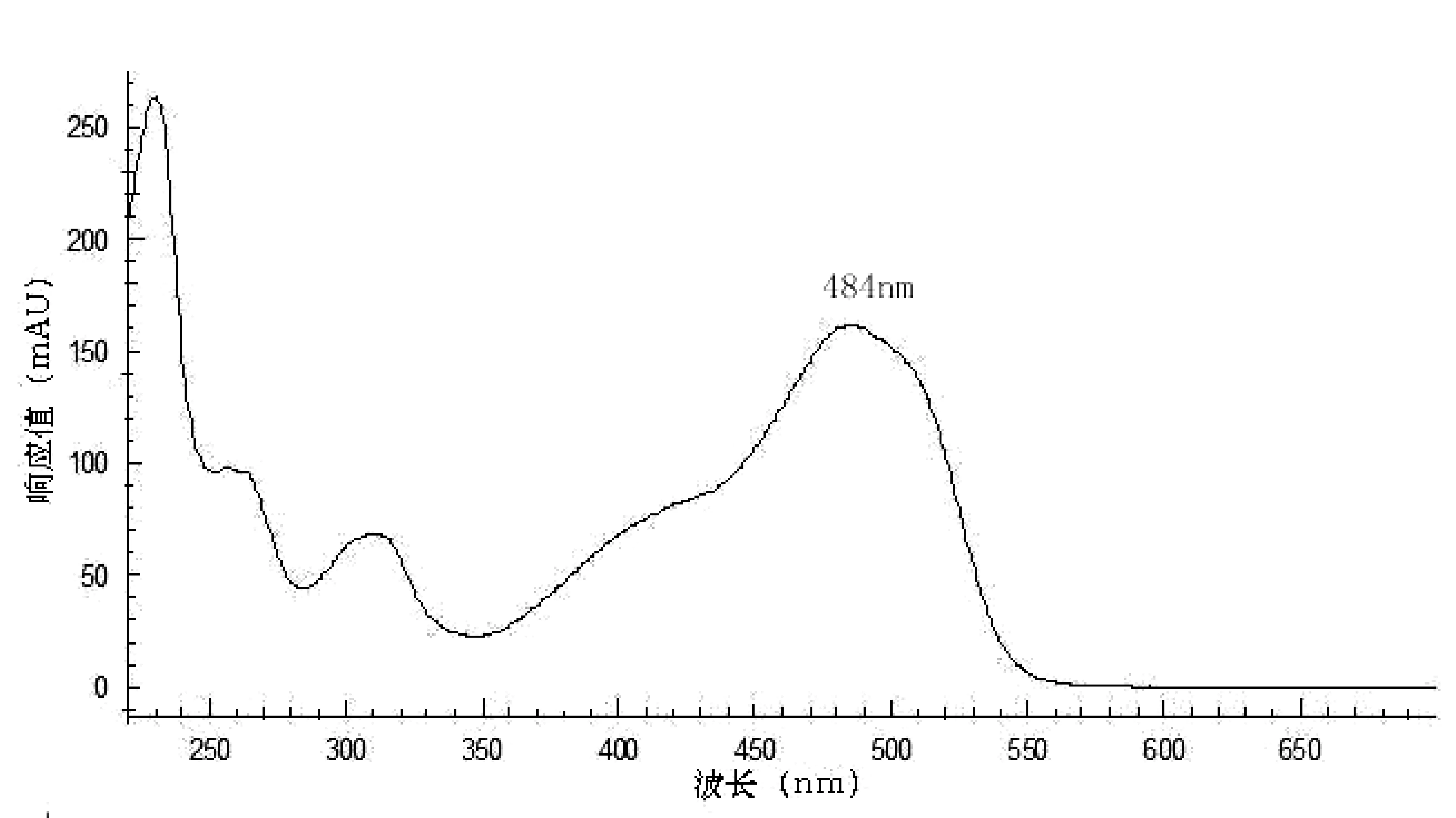
图1 酸性橙Ⅱ的吸收光谱图
2.2 流动相及洗脱方式的选择
试验中采用了甲醇-乙酸铵(80∶20)溶液、甲醇-乙酸铵(15∶85)溶液和甲醇-乙酸铵溶液梯度洗脱3种流动相。结果表明,采用甲醇-乙酸铵(80∶20)溶液洗脱时,出峰时间过短,甚至不出峰,色素和干扰物之间均不能实现很好的分离;采用甲醇-乙酸铵(15∶85)溶液洗脱时,出峰时间过长,峰型较宽与干扰物难以分离;采用甲醇-乙酸铵溶液梯度洗脱,从图2中可以看出效果理想,基线稳定,灵敏度高,峰型尖锐,杂质干扰少。因此,本实验选择甲醇-乙酸铵溶液梯度洗脱方式。
图2 酸性橙Ⅱ标准溶液的液相色谱图(浓度为5.0 μg/mL)
2.3 抗干扰实验
考虑到样品中的常见添加剂苯甲酸、山梨酸、糖精钠、安赛蜜等(1mg/mL),对上述添加剂进行全扫描,上述物质在可见波长下无响应,不干扰测定(图3)。向样品中分别按通常使用量加入常用人工合成食用色素柠檬黄、日落黄、苋菜红、胭脂红等,均不干扰测定(图4)。

图3 苯甲酸、山梨酸、糖精钠、安赛蜜混合紫外-可见扫描图
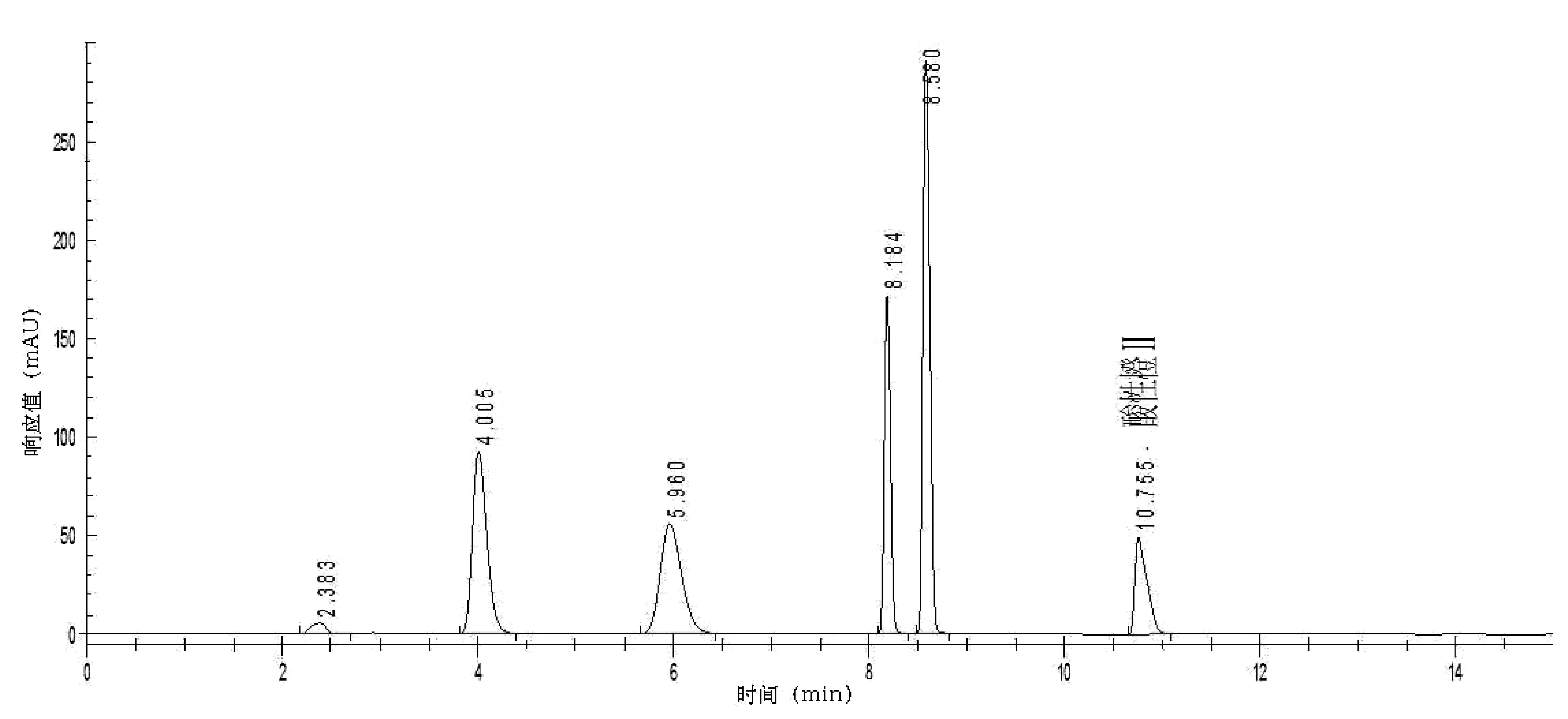
图4 柠檬黄、日落黄、苋菜红、胭脂红及酸性橙Ⅱ混合色谱图
2.4 线性关系及检测限
酸性橙Ⅱ标准工作液系列,按方法的实验条件进样,以所得峰面积A对浓度C(μg/mL)进行回归分析,结果表明在0.1μg/mL~20 μg/mL的标准溶液浓度范围内,酸性橙Ⅱ浓度与响应值有良好的线性关系(图5),其回归方程为:y=34.94x-2.697;相关系数:r=0.9998。根据色谱响应值S/N≥3标准计算,酸性橙Ⅱ检出限为0.08 mg/L。
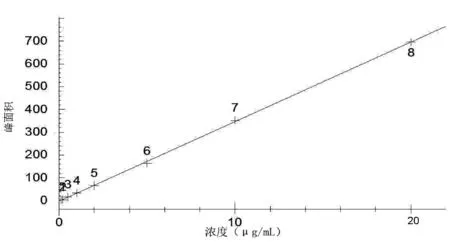
图5 酸性橙Ⅱ标准工作曲线图
2.5 精密度实验
以酸性橙Ⅱ标准溶液5.0 μg/mL,连续进样6次,每次10 μL,测得酸性橙Ⅱ平均峰面积为162.17,RSD为0.13%。
2.6 稳定性实验
以标准溶液5.0 μg/mL,每隔2 h进样,每次10 μL,测得酸性橙平均峰面积为164.57,RSD为0.150%。结果表明:供试品溶液在12 h内所测得的结果基本一致。
2.7 重复性实验
取同一黄鱼样品6份,按1.4.2的方法制成供试溶液。在上述色谱条件下,测定酸性橙Ⅱ(图4)平均含量为4.45 mg/kg,RSD为1.54%。
2.8 回收率实验
称取已知酸性橙Ⅱ含量(4.45 mg/kg)的阳性样品9份,分别作5.00 mg/kg、10.00 mg/kg、15.00 mg/kg 3种浓度的加标回收实验,按1.4.2的方法制成供试品溶液,并按上述色谱条件测定碱性橙的含量,计算回收率(表2)。结果表明,这3种浓度的加标回收率在97.4%~99.2%之间,RSD在3.39%~4.72%之间。
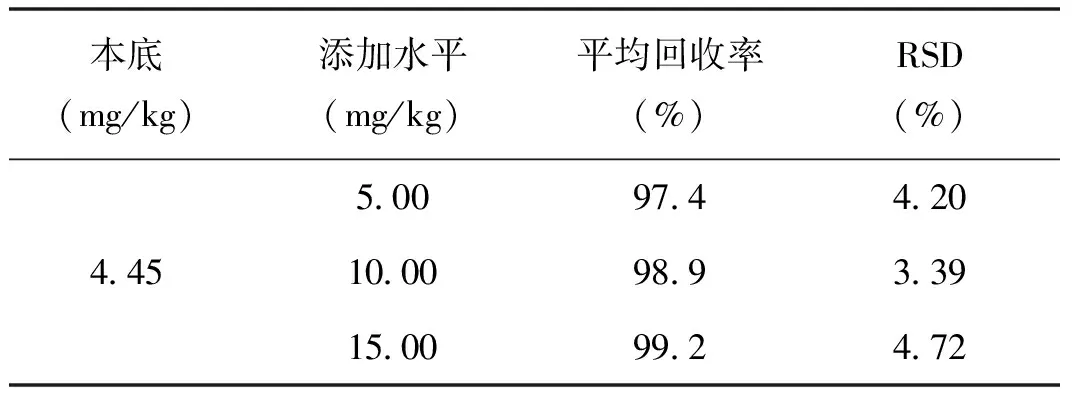
表2 阳性样品中性橙Ⅱ的添加回收率、精密度(n=3)
另取一阴性样品(图5)进行低浓度的加标回收实验,结果见表3。该阴性样品中添加水平为0.50 mg/kg、1.00 mg/kg、2.00 mg/kg,回收率在95.6%~97.3%之间,RSD在4.32%~7.31%之间。
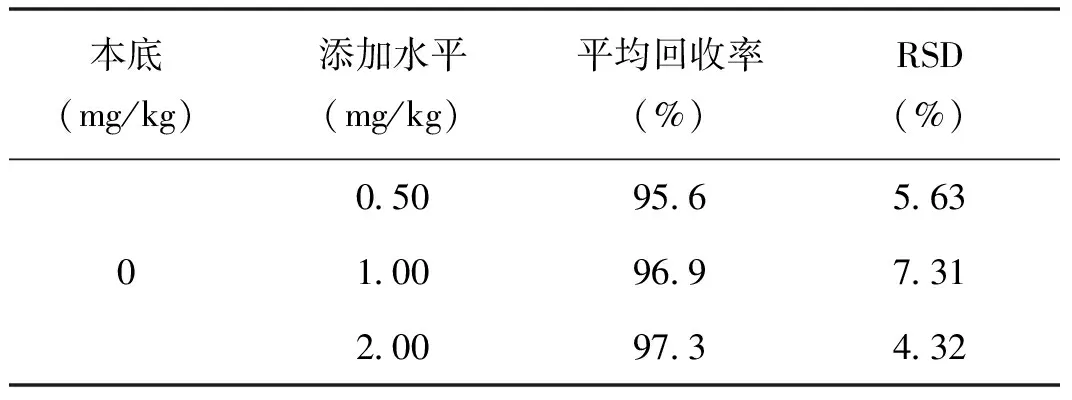
表3 空白样品中性橙Ⅱ的添加回收率、精密度(n=3)
3 结论
本方法操作简便,精密度高,回收率好,在0.1μg/mL~20 μg/mL的浓度范围内线性关系良好,为酸性橙Ⅱ标准方法制定提供理论支持及市场上染色黄鱼制品的卫生监督提供较好的判定依据。
[1]陈大义.纸层析定性示波极谱法测定非食用色素酸性橙[J].中国卫生检验志,2001,11(2):170-171.
[2]雷波.分光光度法对混合染料浓度的同时测定[J].染整技术,2003,25(3):35-37.
[3]张秀尧,蔡欣欣.反相高效液相色谱法快速测定食品中违规使用的酸性橙Ⅱ染料[J].中国卫生检验杂志,2004,14(2):204.
[4]刘华良,荣维广,王联红,等.液相色谱串联质谱法检测鲍鱼汁中的酸性橙Ⅱ[J],中国食品卫生杂志,2010,22(1):16-18.
[5]肖白曼,刘宁,朱正浩.HPLC法测定食品中非食用色素酸性橙Ⅱ[J].中国食品卫生杂志,2005,17(5):422-424.
[6]谢维平,欧阳燕玲,黄盈煜,等.超高效液相色谱法同时测定食品中4种工业染料[J].中国食品卫生杂志,2012,24(4):329-332.
[7]李亚森,潘英,陈艳.用HPLC法测定食品中酸性橙Ⅱ和酸性金黄的探讨[J].职业与健康,2008,24(2):123-124.
[8]陈春晓,刘红河,钟岳桐,等.食品中酸性橙Ⅱ的HPLC-MS/MS测定方法研究[J].中国卫生检验杂志,2008,18(11):2209-2211.
Determination of acid orangeⅡin yellow croaker by HPLC.
Feng Xiaoqing1,Liu Hualiang2,Ruan Liping2, Wang Qin1, Wang Lu1,Wang Yi1,Hang Xueyu1
(1.HuaianCenterforDiseaseControlandPrevention,Huaian223001,China;2.JiangsuCenterforDiseaseControlandPrevention,Nanjing210009,China)
A method for determination of acid orangeⅡ in yellow croaker, was developed.Samples were homogenized, 2 grams of sample were extracted with ethanol for 5 minutes. The extract was separated and purified by polyamide solid phase extraction column. The HPLC separation was achieved by using C18(250 mm×4.6 mm,5μm) with a mobile phase consisting of methanol and acetate buffer, and the gradient program was used at a flow rate of 1 ml/min.The column temperature was set at 30 ℃ and the injection volume was 10 μl. The detection wavelength was at 485 nm for acid orangeⅡ, then we selected the best chromatograph conditions to determine the concentration of acid orangeⅡ in yellow croaker and its added standard recoveries. This method exhibited a linear relation within 0.1-20 μg/ml acid orangeⅡ content. The regression equation was y=34.94x2.697 ( r=0.9998) and the minimum detectable limit of this method was 0.08μg/ml. The recoveries were 96%-99%, the relative standard deviations(RSDs) were 3.39%-7.31% while the contents of added standard were 0.5-10 mg/kg. The method is simple, accurate and reliable.
acid orangeⅡ; yellow croaker; HPLC
冯晓青, 男 ,技师 ,研究方向为卫生检验,Email:fengxiaoqing-20@163.com.。
杭学宇 ,男 ,副主任技师 ,研究方向为理化检验,Email:664686545@qq.com。
10.3936/j.issn.1001-232x.2015.04.004
2015-01-19
猜你喜欢
红蜻蜓·低年级(2024年4期)2024-05-13 06:41:29
读者·原创版(2022年2期)2022-11-09 12:28:03
读者·原创版(2022年2期)2022-02-22 09:23:54
云南化工(2021年10期)2021-12-21 07:33:28
食品安全导刊(2020年21期)2020-09-07 09:14:04
农家科技中旬版(2019年9期)2019-10-08 05:27:47
山西农业科学(2019年6期)2019-06-19 07:14:40
福建基础教育研究(2019年8期)2019-05-28 08:39:51
中学生数理化·八年级物理人教版(2017年6期)2017-11-09 06:00:43
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:52
