
酶的研究与生命科学(三):分子生物学酶的发现和应用
2015-05-25 04:16郭晓强
自然杂志 2015年5期
郭晓强
石家庄职业技术学院化学工程系,石家庄 050081
酶的研究与生命科学(三):分子生物学酶的发现和应用
郭晓强†
石家庄职业技术学院化学工程系,石家庄 050081
20世纪下半叶,分子生物学取得迅猛发展,分子生物学酶的发现和应用在其中发挥了巨大的推动作用。DNA聚合酶、RNA聚合酶、逆转录酶、限制性内切酶和端粒酶等的鉴定和功能阐明拓展了对许多生命现象的理解和认识。这些酶的应用还衍生出重组DNA、桑格酶法测序和聚合酶链式反应等技术,在基因操作、DNA测序和扩增等方面具有广泛应用。通过介绍分子生物学酶的研究历程展现了酶的发现和应用对当代生命科学研究仍有重要意义。
分子生物学酶;DNA聚合酶;逆转录酶;限制性内切酶;分子生物学技术;诺贝尔奖
繁殖是生命的另一个典型特征,而繁殖的基础在于遗传物质的稳定传递。孟德尔豌豆杂交实验开启了遗传学时代,到20世纪中叶遗传学研究开始进入分子水平。1953年,沃森和克里克的DNA双螺旋模型的提出标志着分子生物学的诞生。60多年来,分子生物学领域取得了一系列重大的成果,极大地拓展了人们对生命现象的理解和认识,相关知识的应用也对改善人们的生活质量产生了积极影响。分子生物学研究的中心内容是DNA、RNA及蛋白质的信息传递和相互作用,而酶在其中发挥了关键性作用。从20世纪50年代开始先后鉴定出DNA聚合酶、RNA聚合酶、DNA连接酶、引物酶、逆转录酶、限制性内切酶、拓扑异构酶和端粒酶等,这些酶的研究和应用还推动分子生物学相关技术的发展,诞生了基因重组技术、DNA酶法测序和聚合酶链式反应等多项实验室常用方法。本文通过介绍分子生物学酶的研究历程从另一方面展现分子生物学的发展轨迹。
1 多核苷酸磷酸酶的发现
1953年后,DNA和RNA这两类生物大分子的重要性开始引起科学界的关注,生物化学研究领域也逐渐从物质代谢转移到分子生物学。不久西班牙裔美国生物化学家奥乔亚(Severo Ochoa)就率先取得重大突破,鉴定出多核苷酸磷酸酶。
奥乔亚在西班牙马德里大学医学院获得医学博士学位,早期主要研究肌肉生理学和生物化学,阐明了糖酵解的多步酶促反应。1941年,奥乔亚来到美国,先后在华盛顿大学医学院和纽约大学医学院工作,期间主要对物质代谢特别是三羧酸循环的阐明发挥了关键性作用。他的实验室掌握了酶的鉴定、纯化和分析等技术,奥乔亚本人也成为酶学领域的核心人物之一。DNA和RNA作为遗传物质(RNA病毒)地位的确立使奥乔亚决定从营养物质代谢转向RNA合成研究。
1955年,奥乔亚的一名博士后在研究氧化磷酸化机制过程中,观察到醋酸菌提取液中具有磷酸和ADP交换活性。随后他将负责催化该反应的酶进行分离和纯化,发现该酶可催化二核苷酸(NDP)合成类似RNA分子的多聚核苷酸。由于该酶与多糖合成酶催化功能类似,因此命名为多核苷酸磷酸酶(polynucleotide phosphorylase,PNPase)(图1)[1]。一开始,奥乔亚希望多核苷酸磷酸酶就是负责细胞内RNA生物合成(DNA转录或RNA复制)的酶,但随后发现该酶发挥催化作用时不需DNA或RNA模板,这意味着该酶不符合预期目标。但不久,奥乔亚发现该酶可用于特定多核苷酸链的合成和末端磷酸同位素标记,而这些产物在体外转录实验和遗传密码破译等研究中具有广泛应用。借助多核苷酸磷酸酶合成的寡聚核苷酸,奥乔亚确定了蛋白质翻译过程中RNA方向为5′到3′,并破译部分密码子。多核苷酸磷酸酶是第一个发现的与RNA合成相关的酶,尽管并不负责转录,却激发了多名科学家的灵感以寻找更多核酸生成相关酶。不久奥乔亚的学生美国生物化学家阿瑟•科恩伯格(Arthur Kornberg)就发现了DNA聚合酶。

图1 奥乔亚和多核苷酸磷酸酶
2 DNA聚合酶的发现
阿瑟•科恩伯格在纽约城市学院完成学业。1946年他来到纽约大学医学院奥乔亚实验室接受了系统的酶学训练,随后开始对辅酶和无机焦磷酸酶合成机制进行研究,并发现了核苷酸辅酶如FAD、NAD等的生物合成过程。1953年,科恩伯格加入华盛顿大学。随着DNA重要性的凸显,他将DNA代谢相关酶作为重要研究目标,先后阐明嘌呤和嘧啶核苷酸合成途径的关键步骤。对核苷酸的研究很自然就使科恩伯格想到这些核苷酸如何连接形成DNA,因为双螺旋模型尽管预示DNA具有自我复制能力,却一直未有实验证实。
1955年春,科恩伯格以大肠杆菌提取液为材料,用放射性同位素标记核苷酸的方法证明存在催化脱氧核苷酸多聚化酶。随后科恩伯格决定和学生及同事将该酶纯化,以进一步阐明其生物学活性及催化特性。经过一年多艰苦努力,科恩伯格等终于在1957年将该酶纯化。在该酶的催化作用下以四种脱氧核苷三磷酸(deoxynucleoside triphosphate,dNTP)为原料可以合成DNA,并且需要DNA作为模板(图2)[2-3]。需要指出的是,科恩伯格使用该酶首次在试管中合成DNA,这是生命科学史上的一个重大进步,说明DNA可在DNA聚合酶催化下合成新DNA链,即DNA复制,而DNA复制是物种遗传信息传递的基础。

图2 科恩伯格和DNA聚合酶
1959年,奥乔亚和科恩伯格由于“发现RNA和DNA的生物合成机理”而分享诺贝尔生理学或医学奖。值得一提的是,奥乔亚的获奖是一个“错误”,因此阿瑟•科恩伯格在后来一篇回忆文章中认为颁奖词应纠正为“发现RNA样聚合物和DNA的生物合成机理”更为符合[4]。由于多核苷酸磷酸酶在遗传密码破译中的重要性,奥乔亚获奖也算实至名归。其实,阿瑟•科恩伯格的发现后来证明也存在一定问题。他发现的酶被命名为DNA聚合酶Ⅰ,但该酶并非负责细菌内DNA复制,而主要参与DNA损伤修复。真正发挥DNA复制的酶为DNA聚合酶Ⅲ,该酶由阿瑟二儿子托马斯(Thomas Bill Kornberg)于1972年鉴定成功,也算“自我”弥补。
诺贝尔奖出现“失误”的一个重要原因在于DNA和RNA生物合成在当时的重要性。奥乔亚的发现间隔4年,而阿瑟的发现只有1年多时间,充分体现了科学界对这两项工作的高度认可(DNA双螺旋模型的获奖还等了9年),同时也说明酶的重要性。DNA聚合酶的发现意义重大,一方面初步阐明DNA的复制机制,深化对生命过程的理解和认识,另一方面还为分子生物学提供重要工具。DNA聚合酶在多种技术如DNA重组、DNA测序、聚合酶链式反应(polymerase chain reaction,PCR)等都具有广泛应用。
3 RNA聚合酶结构的阐明
真正催化细菌DNA转录的RNA聚合酶于1959年由美国生物化学家赫尔维茨(Jerard Hurwitz)等发现。10年后美国生物学家卢德(Robert Roeder)进一步鉴定出三种真核RNA聚合酶,它们分别负责不同RNA(rRNA、mRNA和tRNA等)前体分子的转录,从而全面开启转录机制的研究。20世纪70和80年代,科学家还先后鉴定出多种通用转录因子和特异性转录因子,它们和RNA聚合酶共同参与转录完成,但转录的详细分子机制则由美国结构生物学家罗杰•科恩伯格(Roger David Kornberg)阐明。
罗杰是阿瑟的大儿子,在哈佛大学获得化学学士学位,后进入斯坦福大学进行结构生物学研究。20世纪70年代,罗杰来到当时结构生物学的“圣地”——英国剑桥大学分子生物学MRC实验室进行博士后研究,并于1974年发现核小体结构。
1978年,罗杰加入斯坦福大学,开始了对真核生物转录的机理研究。为了简化实验,罗杰放弃使用小鼠和其他哺乳动物为研究材料,而是选择较简单的酵母。三种RNA中,编码蛋白质的mRNA种类最为多样,转录调节也最为精细,因此罗杰将RNA聚合酶Ⅱ作为研究对象。罗杰首先成功制备出酵母体外转录体系,从中发现参与mRNA转录的第三大类元件——中介体(mediator),它们与RNA聚合酶Ⅱ和转录因子构成转录器。
20世纪90年代,在了解真核生物转录基本元件及功能基础上,罗杰决定确定基本元件间相互作用及调节转录的机制,这需要借助结构生物学手段来实现。真核转录复合物由多亚基构成,以较简单的酵母为例,其RNA聚合酶有12个亚基,转录因子26个亚基,而中介体21个亚基,因此形成一个拥有近60个亚基、分子量超过3 000 kD的大转录器(图3)。此外,研究面临的难题还包括多蛋白复合物稳定性差,而经典的结构研究方法重点在静态结构,因此对转录动态变化的研究贡献有限。在随后十几年中,多个研究小组都投入巨大精力,但进展缓慢。罗杰凭借X射线晶体衍射方面熟练的技巧和研究过程中的改进,实现了将电子衍射技术和脂质层技术相结合的方法,从而获得二维蛋白晶体等。
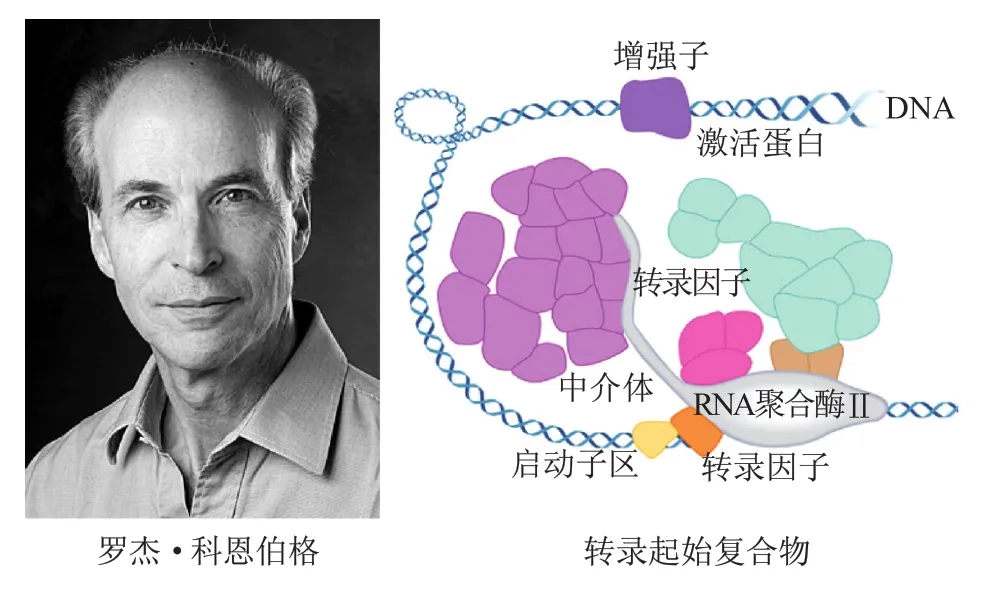
图3 罗杰和RNA聚合酶
2001年,罗杰的研究取得重大突破,第一次在分子水平上阐明了转录机制。罗杰获得酵母RNA聚合酶Ⅱ在2.8A°分辨率上的结构[5],发现RNA聚合酶Ⅱ的两个最大亚基位于DNA结合部位两侧,而其他小亚基则位于外围。随后几年,罗杰又相继获得许多转录过程复合物的结构,从而对转录RNA聚合酶Ⅱ、模板DNA、新形成RNA、四种核苷酸及转录因子在转录过程中的位置和作用方式等有了一个清晰、动态的理解,对DNA转录过程中的移位、链分离、核苷酸选择、启动子识别等过程也有了更深一步的认识。由于RNA聚合酶及相关亚基在酵母和高等哺乳动物(包括人)中的高度保守性,这些研究将对理解人类基因的转录机制具有重要益处。
2006年,罗杰由于“真核细胞转录分子基础方面的研究”而获得诺贝尔化学奖。转录是基因表达的关键阶段,其精细调节对保证机体正常状态具有十分重要的意义,如干细胞的增殖和分化,器官的发育和形成等。基因转录调节异常可导致肿瘤、炎症和代谢性疾病等的发生。因此,对转录过程精细机制的理解具有重大的理论和应用价值。
4 逆转录酶的发现
美国生物化学家特明(Howard Martin Temin)小时候就对科学表现出极大兴趣,在宾夕法尼亚的斯沃斯茅学院完成生物学教育后进入加州理工学院进行博士研究,跟随意大利裔美国科学家杜尔贝科(Renato Dulbecco)探索病毒的致癌机制。杜尔贝科研究可引发小鼠肿瘤的多瘤病毒(DNA病毒),发现病毒DNA进入宿主细胞后可插入宿主DNA,从而作为整体一起复制,宿主由于获得病毒基因而出现癌变。然而,特明选择的是劳斯肉瘤病毒,这是一种RNA病毒,它如何诱导细胞癌变成为需要解决的难题。在研究中特明发现,尽管劳斯肉瘤病毒遗传物质是RNA,但抑制DNA复制也影响病毒复制;因此特明于1965年根据杜尔贝科理论提出解释劳斯肉瘤病毒致癌机制:RNA病毒进入宿主细胞后,首先将RNA转化为DNA,生成的DNA再与宿主细胞DNA整合,随宿主一起复制和遗传。
特明理论很好地解释了RNA病毒的致癌机制,却遭到许多科学家的反对。按照1958年克里克描述信息传递的“中心法则”,遗传信息一般从DNA传递给RNA,而不逆行。而且特明理论中的一个关键问题是RNA如何将信息传递给DNA尚未用实验证实。随后几年中,特明和同事以劳斯肉瘤病毒为材料寻找具有催化RNA到DNA的酶。
1970年,特明鉴定出以RNA为模板的DNA聚合酶[6],由于该酶催化的反应与转录过程(以DNA为模版合成RNA)正好相反,因此将该酶称为逆转录酶。与此同时,特明的校友美国科学家巴尔的摩(David Baltimore)也鉴定出逆转录酶,进一步证实了特明的发现。
巴尔的摩也毕业于斯沃斯茅学院,接受了较系统的分子生物学培训。1964年巴尔的摩从麻省理工学院获得生物物理博士学位,之后不久获悉特明提出的RNA病毒致癌假说。尽管大多数科学家都持怀疑态度,但巴尔蒂摩敏锐地察觉到该理论的重要性,并认为自然界一定存在该过程,因此决定将研究方向转向病毒领域。

图4 特明、巴尔的摩与逆转录酶
1965年,巴尔的摩进入索尔克研究所与杜尔贝科合作研究病毒学,不久从脊髓灰质炎病毒(RNA病毒)鉴定出一种RNA聚合酶,但随后结果令巴尔的摩失望。该酶是以RNA为模板,因此并不符合特明假说。1968年,巴尔的摩回到麻省理工学院,继续进行病毒的研究。功夫不负有心人!1970年,巴尔的摩凭借酶学研究娴熟的技术最终从可诱发癌变的鼠白血病病毒中鉴定出RNA依赖的DNA聚合酶[7]。逆转录酶可首先以RNA为模板合成出互补DNA(cDNA),随后将模板RNA降解,接下来再以cDNA为模板合成另一条DNA链而生成双链DNA(图4),双链DNA可整合到宿主DNA而实现致癌。
1975年,特明和巴尔的摩以及杜尔贝科由于“肿瘤病毒和细胞遗传材料间相互作用的发现”而分享诺贝尔生理学或医学奖。当初颁奖重点在于病毒的致癌机制,然而令大家意想不到的是随后的发现证明逆转录酶更为重要。逆转录现象和逆转录酶的发现是现代医学最重要的发现之一,它促使克里克1970年修改中心法则,还推动逆转录病毒的研究,如导致艾滋病的HIV的发现等。另外,逆转录酶还成为分子生物学研究必不可少的基本工具,如DNA探针制备,基因工程中使用mRNA制备cDNA等。
至此,中心法则相关的三种酶——复制需要的DNA聚合酶、转录需要的RNA聚合酶以及逆转录涉及的逆转录酶都被授予诺贝尔奖。此外,分子生物学技术相关酶类也被鉴定成功。
5 限制性内切酶的提出、分离和应用
瑞士科学家阿尔伯(Werner Arber)在苏黎士联邦理工学院获得学士学位,随后在日内瓦大学完成生物物理学博士学习,师从著名的生物物理学家威格尔(Jean-Jacques Weigle)研究噬菌体。20世纪50年代,科学家发现一种λ噬菌体对不同的大肠杆菌菌株具有不同的感染能力,如极易感染大肠杆菌C菌株的λ噬菌体对K菌株的感染力只是C菌株的千分之一,因此将大肠杆菌K菌株限制λ噬菌体感染的能力称为“宿主控制的噬菌体限制”。这种现象引起阿尔伯极大的兴趣。
1960年,阿尔伯加入日内瓦大学,着手全面研究噬菌体限制的分子基础。1962年,阿尔伯和研究生发现感染λ噬菌体并形成溶源状态的大肠杆菌K12(K12-λ)对野生型λ噬菌体再次感染具有抵抗作用,但对溶菌后释放的λ噬菌体(λ-P1)无抵抗作用,考虑到两种噬菌体DNA序列一致,因此可能变化在于DNA结构方面(图5)。根据这种现象,阿尔伯推测宿主细胞中存在一种核酸内切酶,可将侵染的噬菌体DNA切割成特定片段,从而实现限制作用;同时宿主细胞还存在DNA甲基化酶,以对自身DNA(包括溶源化后整合的λ噬菌体DNA)修饰以避免核酸内切酶的切割[8-9]。尽管阿尔伯还没有发现这样的酶,但推测该内切酶具有识别特定核苷酸序列的能力,并在特定位置实现DNA切割。阿尔伯将这种双酶理论称为限制-修饰系统,这种理论不久就得到证实。1965年,阿尔伯发现噬菌体特定核苷酸序列的突变可造成其对限制系统不敏感,而宿主细胞感染噬菌体后确实存在DNA甲基化修饰。1968年,阿尔伯从大肠杆菌中鉴定出一种内切酶EcoB,另一小组获得EcoK。这两种酶都可识别特定核苷酸序列,但在随机位置对DNA切割不具特异性,因此并非阿尔伯推测的酶。后来将其称为Ⅰ型限制性内切酶。1970年,具有识别和切割双重特异性的Ⅱ型限制性内切酶被曾经与阿尔伯一起工作过的美国微生物遗传学家史密斯(Hamilton Othanel Smith)发现。
史密斯从加州大学伯克利分校获得生物学学士学位,后又从约翰霍普金斯医学院获得医学学位,但最终未成为一名医生,而成为了一名科学家。史密斯很早就对细菌遗传学情有独钟,然而在研究细菌遗传重组时却无法获得噬菌体DNA,这个现象使他联想到限制现象。1966年,史密斯来到瑞士日内瓦阿尔伯的实验室,更加熟悉了阿尔伯限制-修饰假说,同时对限制酶有了初步认识。
回到美国后,史密斯全身心投入限制酶的分离工作。1970年,史密斯和同事经过艰苦努力后终于从嗜血流感细菌(Haemophilus influenzae)中分离得到一种内切酶。该酶可特异性识别双链而不是单链DNA,并且只对噬菌体(外源)DNA具有切割作用,而对细菌DNA无任何影响。该酶对双链DNA的切割不产生游离核苷酸,因此具有核酸内切酶活性。由于所获得酶切片段较大,因此切割具有位点特异性(图5)[10],这正符合阿尔伯所预测的限制性内切酶。史密斯和同事还鉴定了该酶的识别及切割序列(5′GTT/C↓G/AAC 3′),该酶被命名为HindII,这是人类发现的第一个Ⅱ型限制性内切酶。限制性内切酶的命名原则是前三个字母代表种属名,第四个字母为株名,而最后数字表示鉴定的先后顺序。如HindII就表示嗜血流感细菌(H和in)d株第二个酶。HindII的发现引起科学界极大关注,从而掀起一场寻找新型、可用的Ⅱ型限制性内切酶热潮,而另一方面限制性内切酶巨大的应用潜力不久就被史密斯同事内森斯(Daniel Nathans)发现并首先应用。
内森斯从特拉华大学获得学士学位,随后又从华盛顿大学获得医学博士学位。然而在工作中,他却对基础科学产生浓厚兴趣,开始病毒研究,主要是关于病毒与宿主的控制关系。20世纪60年代,科学家将病毒研究从细菌病毒(噬菌体)转向哺乳动物病毒,而内森斯选择猿猴病毒40(Simian virus 40,SV40)为材料研究其致癌机制,却由于缺乏有效的实验手段而进展缓慢。1969年春,身在以色列进行研究的内森斯接到同事史密斯的来信,告诉他发现了Ⅱ型限制性内切酶HindII,该酶可在特定位置切割DNA。在获悉限制性内切酶鉴定成功后,内森斯兴奋异常,立刻意识到这些酶可像胰蛋白酶或胰凝乳蛋白酶作用于蛋白质一样实现对DNA的特异性剪切,从而将DNA消化为特定片段。对于SV40而言,通过限制性酶切而获得小片段DNA,再通过对这些小片段DNA的定位和功能研究而确定病毒的致癌机制。
1969年秋,内森斯回到约翰霍普金斯大学。首先,他用限制性内切酶HindII对SV40的DNA进行酶切,将产物进行放射性同位素标记后用聚丙烯酰胺凝胶电泳进行分离,曝光结果显示共获得大小不同的11个片段(A到K,字母根据片段大小命名,其中A最长)[11],并确定这些片段在SV40中的相应位置(图5)。随后,内森斯又应用其他Ⅱ型限制性内切酶对SV40进行切割,一方面获得了更清晰的限制性内切酶图谱,为深入理解基因结构提供重要信息,另一方面通过分离研究小片段DNA而阐明相关基因的生物学功能。

图5 阿尔伯、史密斯和内森斯与限制性内切酶
1978年,阿尔伯、史密斯和内森斯由于在“限制性核酸内切酶的发现及其在分子遗传学中的应用”而分享诺贝尔生理学或医学奖(图5)。限制性内切酶的发现及应用开启了遗传学研究的新纪元。限制性内切酶为生命科学研究提供了重要工具,如DNA测序、基因工程等。不夸张地说,没有限制性内切酶的应用就没有基因工程的出现。限制性内切酶可将大片段DNA剪切为小片段,从而大大促进高等生物的遗传学研究,人类基因组计划的实施也得益于此(DNA限制性图谱是其中的关键性环节)。限制性内切酶被称为DNA研究中的手术刀。通过特定限制性酶可实现基因染色体定位,可分析基因的化学结构和调节基因表达的特定区域等。限制性内切酶在临床上也具有巨大应用,为先天性畸形、遗传性疾病及肿瘤的预防和治疗提供帮助。现在临床的许多分子生物学诊断中,限制酶都是必备的工具,并因此成为现代医学中最为基本的应用试剂。限制性内切酶的发现引发了遗传学的一场革命,对推动分子生物学的发展具有不可估量的作用。
6 端粒酶的鉴定
染色体是真核生物遗传信息的载体,其末端的特殊结构端粒(telomeres)对防止不同染色体间末端融合和维持染色体的完整性具有重要意义。早在20世纪30年代,科学家就观察到端粒结构,但对这种结构的特征知之甚少。澳大利亚和美国遗传学家布莱克本(Elizabeth Helen Blackburn)首先在该领域取得突破。
布莱克本在澳大利亚墨尔本大学获得生物化学学位,1975年从剑桥大学获得博士学位(导师是桑格教授)。她在耶鲁大学博士后期间,利用刚刚建立起来的桑格测序法测定了四膜虫的端粒结构,惊奇地发现它是由六核苷酸组成的串联重复序列。1977年,布莱克本加入加州大学伯克利分校继续进行端粒研究,后与哈佛医学院遗传学家绍斯塔克(Jack William Szostak)进一步确定酵母染色体端粒上也存在短重复序列,随后对多种真核生物端粒结构的鉴定说明末端DNA重复是一种普遍现象。接下来的问题是真核生物如何产生端粒,布莱克本认为特定酶催化了该过程,而格雷德(Carol Widney Greider)最终证明该酶的存在。
格雷德从加州大学圣巴巴拉分校获得生物学学位,1984年5月加入布莱克本实验室开始博士学习,并对端粒延长机制产生了浓厚兴趣。格雷德首先利用生物化学方法鉴定是否存在具有催化端粒序列延长的酶。由于对该酶的特性一无所知,因此只能摸索条件进行。格雷德将通过酶切获得的端粒模板和放射性标记的脱氧核苷三磷酸底物与四膜虫细胞核提取液混合,一段时间后提取DNA,检测其长度变化。不久,格雷德就从电泳结果中发现特异的六核苷酸重复,然而随后发现这是由于DNA聚合酶催化的缘故。在经历多次失败后,1984年底格雷德将原来的端粒模板换成人工合成的短六核苷酸重复,反应完毕后利用电泳检测,结果发现生成一系列相差6个核苷酸的一系列DNA延长产物,这是该酶存在的第一个证据。进一步实验排除了其他已发现酶特别是DNA聚合酶功能的前提下,她们最终确定细胞中存在一种催化端粒末端延长的新酶,将其命名为端粒末端转移酶(telomere terminal transferase)[12],后简称端粒酶(telomerase)(图6)。格雷德和布莱克本深入研究证明端粒酶由RNA和蛋白两部分构成,蛋白部分具有逆转录酶活性,可利用RNA为模板将特定脱氧核苷酸依次添加到染色体末端。

图6 布莱克本、格雷德与端粒酶
科学家进一步对端粒和端粒酶研究后发现,端粒长短与细胞寿命成正相关。随着年龄的增加,端粒酶活性降低从而造成端粒长度变短,细胞出现衰老表型。相反,癌细胞内端粒酶活性持续性激活从而使细胞出现“不死性”。这些发现拓展了人们对衰老和肿瘤等相关疾病的认识,从而为其治疗提供新的疗法。
2009年,布莱克本、绍斯塔克和格雷德由于 “端粒和端粒酶对于染色体保护作用”方面的发现而分享诺贝尔生理学或医学奖。
7 重组DNA技术的实现
酶学发展还直接推动了技术革新,为生命科学的进一步发展提供了重要工具,包括重组DNA技术、DNA酶法测序和聚合酶链式反应等。重组DNA技术是当前生命科学领域最常用的方法之一,为许多问题的解决带来便利,而突破是在20世纪70年代初由美国科学家伯格(Paul Berg)首先完成。
伯格在宾夕法尼亚州立大学完成生物化学的学习后进入俄亥俄州西储备大学进行博士学习,主要进行一碳单位代谢的研究。1953年,伯格进入圣路易斯华盛顿大学,先后研究了脂肪酸代谢、氨基酸活化等过程,鉴定出氨基酰-tRNA及相关酶类。1959年,伯格来到斯坦福大学医学院担任生物化学教授,开始研究细胞内蛋白质合成机制,但研究材料由大肠杆菌转移到哺乳动物细胞,此外还接触到SV40,并决定探索SV40中相关基因的功能。20世纪70年代初,限制性内切酶的发现为分子生物学的研究带来极大便利。伯格基本确定了SV40的基因分布,并测定了部分基因的核苷酸序列,接下来要确切获悉特定的基因功能则需要首先让基因表达出蛋白质。由于SV40病毒侵染宿主细胞后可导致癌变,因此伯格考虑如将两个不同物种间基因整合后再侵染细胞则可减弱致癌效应。
伯格首先利用限制性内切酶将SV40的DNA切割成一些短片段,然后利用末端转移酶在双链分子末端添加碱基,从而形成含有末端延长的双链DNA(该末端被称为粘性末端)。伯格又用相同方法对λ噬菌体DNA进行操作,从而也获得拥有末端延长的噬菌体DNA,且两端添加的碱基恰好与SV40末端碱基互补配对。1972年,伯格和同事将两者混合并最终获得含有两个物种DNA的杂合分子,第一次实现了体外DNA重组(图7)[13]。

图7 伯格和DNA重组技术
1980年,伯格由于“在核酸生物化学方面的基础性贡献,特别是体外重组DNA的实现”而分享诺贝尔化学奖的一半。重组DNA技术开创了遗传工程的先河,一年后就实现了基因工程。基因工程可打破物种限制,理论上可将任何物种间基因实现有效整合,对分子生物学的发展具有革命性影响。重组DNA技术一方面为基因操纵提供了一种强有力的工具,在基因结构与功能、基因表达调节和转基因研究等方面发挥了重要作用;另一方面还具有重要的医学应用价值,科学家利用简单生物如细菌作为宿主来表达大量感兴趣的目的蛋白,如胰岛素、生长素、干扰素等。今天基因工程药物的商业应用已成为一个巨大且具有重大潜力的生命科学领域,是许多药物和其他化学药品的工业基础。此外,重组DNA技术还为转基因技术和基因治疗等提供了重要手段。
8 DNA酶法测序的发明
桑格(Frederick Sanger)在剑桥大学完成化学学习,从20世纪40年代开始进行胰岛素化学结构的研究,最终于1955年确定胰岛素中氨基酸的排列顺序,并因此荣获1958年诺贝尔化学奖。50年代,桑格成为剑桥医学研究委员会分子生物学实验室成员。60年代开始,桑格开始进行RNA测序研究,采用类似蛋白质测序基本原理——部分降解法。首先用特定核酸酶对RNA在特定碱基处切割,然后纯化短片段RNA,进一步利用化学降解确定小片段顺序,最终通过重叠法推导出全长RNA核苷酸序列。1967年,桑格小组利用这种方法确定了大肠杆菌5S rRNA序列(120个核苷酸构成)。部分降解法涉及到连续降解和分离过程,因此整个过程相当缓慢和繁琐。尽管有科学家用这种方法进行DNA测序,但是一般只能达到50个碱基,测定更长DNA则显得困难重重。然而,作为遗传物质的DNA一般都有几千甚至上万碱基对,因此完成这项任务更是遥不可及。随着DNA的重要性越来越得到科学界的认可,DNA测序也就自然成为分子生物学领域需要迫切解决的关键问题之一,而传统方法的弊端使科学界开始寻找新的思路,桑格将注意力转向了复制程序。
DNA聚合酶可利用DNA单链为模板,通过碱基配对获得互补链,分析互补链的碱基序列可反推原DNA序列。桑格发现核苷三磷酸可代替脱氧核苷三磷酸掺入互补链,形成的互补链可被强碱试剂在核苷酸处断裂,但对脱氧核苷酸无影响。根据添加到反应体系中核苷三磷酸类型(如ATP),就可确定断裂位置碱基为A,原模板对应位置为T。这种改进较传统方法有很大提高,但整个过程涉及大量分离与分析操作,仍然无法有效应用于DNA测序。
1975年,桑格和库尔森(Alan Coulson)改进测序程序,提出了“加减法”[14]。基本程序:首先在反应体系中加入待测DNA模板、合适引物、四种同位素标记的脱氧核苷三磷酸(dNTP)和DNA聚合酶,反应一段后去除未反应dNTP,将剩余部分进行随后的加法或减法程序。加法程序是将溶液分成四份,分别只加入一种dNTP(如dATP)。由于DNA聚合酶本身的3′→5′外切酶活性,因此对已生成的互补链进行酶切;但由于添加了dATP,因此在碱基A处活性消失,最终获得一系列以A为3′末端的长短不一的片段。将这些片段在丙烯酰胺电泳中根据大小进行分离,从而确定碱基A的位置。同样原理获得另外三种碱基位置。减法程序是将溶液分成四份,然后加入三种dNTP(如缺少dATP)。此时DNA聚合酶催化5′→ 3′延长过程,由于dATP缺乏,因此延长在A前一碱基处停止,最终亦生成大量长短不一的片段。同样利用电泳可确定碱基A位置(相应片段长度加1)。该方法可一次测定80个核苷酸,使用该方法,他们完成了单链噬菌体ΦX174的5386核苷酸测序。这是第一个完成测序的基因组。加减法相对传统方法有极大改善,但仍然非常费力,且误差较多。
1977年,桑格和同事对加减法进行进一步完善,引入双脱氧末端终止法进行测序,也被称为“桑格法”或酶法[15]。双脱氧核苷酸在结构上与脱氧核苷酸类似,但缺失了3′羟基,因此可以和脱氧核苷酸类似被整合到延长的核苷酸链,但由于羟基的缺失而造成进一步延长反应终止。桑格法基本程序:反应体系分为四份,他们共同存在单链模板、同位素标记的互补引物、四种dNTP和DNA聚合酶,然后四份中分别加入双脱氧的A、T、G和C四种核苷酸,最终获得四种分别以A、T、G和C为末端的长短不一序列,利用丙烯酰胺电泳分离后借助放射自显影确定碱基位置,最终确定模板的碱基顺序(图8)。

图8 桑格和DNA测序
桑格的酶法测序是一个巨大突破,可实现快速、精确的长链DNA测序。应用该方法,桑格和同事先后完成人类线粒体DNA(16 569个碱基对)和λ噬菌体(48 502个碱基对)的测序工作。1986年,桑格法被胡德(Leroy Hood)进一步改进,用四色荧光代替同位素并实现测序自动化。这种改进更增强了测序的快速、便捷,在人类基因组测序中发挥了关键性作用。
1980年,桑格由于“核酸碱基序列确定方面的贡献”而与吉尔伯特(Walter Gilbert)(化学法测序发明者)分享诺贝尔化学奖的一半。今天,桑格法测序已成为分子生物学研究必不可少的方法之一,对推动生命科学的发展发挥了关键作用。
9 聚合酶链式反应的发明
DNA是遗传信息的载体,对它的研究成为基础研究和商业生产的基础。然而研究过程中如何得到大量DNA是一个最基本的问题。最早利用大肠杆菌体内扩增技术虽使这一问题得到部分解决,但存在两大弊端,一是耗时,二是需要扩增整个DNA从而增加复杂性,因此无法很好满足实际需要。直到美国工程师穆里斯(Kary Banks Mullis)发明聚合酶链式反应(polymerase chain reaction,PCR)技术才真正解决问题。
穆里斯从小就对科学充满兴趣且极富个性,在高中时就设计成功简易火箭,并携带一只青蛙升高到7 000英尺高空,随后打开降落伞使活的青蛙安全返回地面。穆里斯选择以理工见长的乔治亚理工学院学习化学,在这里建造了一个生产毒药和爆炸物的实验室,而且还发明了由脑电波刺激的电子设施以控制电的开关。1966年他获得化学学士学位,1973年从加州大学伯克利分校获得生物化学博士学位。
由于对大学教育的失望和对未来感到茫然,穆里斯于1979年离开加州大学旧金山分校,进入加州新建的Cetus生物科技公司。该公司主要进行化合物合成,以提供其他科学家进行遗传克隆研究。在工作期间,穆里斯对公司的日常要求感到厌倦,因此花费大量时间在屋顶进行日光浴,还有就是书写计算机程序以能够自动对工作要求进行反应。穆里斯认为真正好的科学发明并非来自于艰苦工作,真正的重大进展往往是由具有好动性格的边缘人士完成的。
1983年,穆里斯乘车去加利福尼亚州西北部门多西诺角农场时,构思了聚合酶链式反应,提出可使用一种随机方式进行遗传物质扩增。当穆里斯将自己的想法介绍给同事时,他们并没有意识到该方法的重要性;而等到Cetus公司意识到该思想的重要性后,该研究就迅速成为公司的中心问题。1985年,穆里斯和同事使用血红蛋白β亚基DNA两端分别互补的两端20个碱基寡核苷酸为引物,通过变性、退火和延伸20个重复反应(图9),最后获得1 μg血红蛋白β亚基DNA扩增片段[16]。最早PCR反应存在两大弊端:一是所用DNA聚合酶是从大肠杆菌中得到的Klenow大片段,具有热不稳定性,高温易变性,且每一周期反应(变性、退火和延伸)都需重新加入一次酶试剂,大大限制该技术的应用;二是缺乏自动化。一开始设置三个不同温度的温箱,根据需要在三个温箱中进行温度变化,因此非常繁琐。1988年使用Tag DNA聚合酶代替Klenow大片段,该酶热稳定强,整个反应只需添加一次即可,这也促进自动化的出现。研究人员又设计出温度调节系统,可预先设置温度,然后在实验过程中根据需要实现调节。随着PCR技术优越性的广泛体现及需求增多,很快成功开发出商业用途的版本。该机器被称为热循环器,只需加入DNA模板、引物、四种核苷酸、DNA聚合酶以及合适的缓冲液,再设定条件就可自动合成一段特异DNA片段。这样的机器经济实惠,即使一个小的实验室也可承担,科学家评论该机器甚至比自然界有更高的复制效率。

图9 穆里斯和PCR
1993年,穆利斯由于“聚合酶链式反应方法”的发明而分享诺贝尔化学奖。PCR的出现在分子生物学、遗传学、医学和考古学引发了一场革命,推动了DNA克隆、DNA测序、基因功能分析等方法学的革新。没有PCR技术,今天人类基因组计划根本无法完成。PCR技术还在进化生物学领域引起了一场革命,可以对古生物遗留的痕量DNA实现扩增,以了解它们的演化过程。此外,PCR技术还在法医鉴定、遗传性疾病诊断、传染性疾病研究等方面发挥关键性作用。
上面重点介绍的分子生物学相关的诺贝尔奖成果已充分体现酶对分子生物学发展的重要性。此外,其他一些成果,如比德尔(George Wells Beadle)和塔特姆(Edward Lawrie Tatum)提出的“一个基因一个酶”(1958年诺贝尔生理学或医学奖)、莫诺(Jacques Lucien Monod)和雅各布(François Jacob)研究β-半乳糖苷酶提出的操纵子假说(1965年诺贝尔生理学或医学奖)、法厄(Andrew Zachary Fire)和梅洛(Craig Cameron Mello)发现的RNA干扰现象中的Dicer酶(2006年诺贝尔生理学或医学奖)中酶也具有重要地位。目前,分子生物学前沿领域如表观遗传学也与酶密不可分。表观遗传学主要涉及DNA甲基化、RNA干扰和组蛋白修饰等,相关酶包括DNA甲基转移酶、组蛋白乙酰转移酶/去乙酰化酶、组蛋白甲基转移酶/去甲基化酶等,在该领域做出卓越贡献的科学家也有望摘取诺贝尔奖。
总之,酶是生命的“媒介”,对生命而言具有至关重要的意义。其在分子生物学发展过程中功不可没,为推动这一领域的快速发展发挥了关键性作用。由于篇幅所限,无法展现60多年来分子生物学酶发展的全貌(如DNA连接酶等发现及应用),只通过一个轮廓性介绍展现酶的重要性,以此纪念DNA双螺旋提出60周年。
(2013年10月14日收稿)
[1]GRUNBERG-MANAGO M, ORITZ P J, OCHOA S. Enzymatic synthesis of nucleic acidlike polynucleotides [J]. Science, 1955, 122(3176): 907-910.
[2]LEHMAN I R, BESSMAN M J, SIMMS E S, et al. Enzymatic synthesis of deoxyribonucleic acid. I. Preparation of substrates and partial purification of an enzyme from Escherichia coli [J]. J Biol Chem, 1958, 233(1): 163-170.
[3]BESSMAN M J, LEHMAN I R, SIMMS E S, et al. Enzymatic synthesis of deoxyribonucleic acid. II. General properties of the reaction [J]. J Biol Chem, 1958, 233(1): 171-177.
[4]KORNBERG A. Remembering our teachers [J]. J Biol Chem, 2001, 276(1): 3-11.
[5]CRAMER P, BUSHNELL D A, KORNBERG R D. Structural basis of transcription: RNA polymerase II at 2.8 angstroms resolution [J]. Science, 2001, 292: 1863-1876.
[6]TEMIN H M, MIZUTANI S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus [J]. Nature, 1970, 226(5252): 1211-1213.
[7]BALTIMORE D. RNA-dependent DNA polymerase in virions of RNA tumour viruses [J]. Nature, 1970, 226(5252): 1209-1211.
[8]ARBER W, DUSSOIX D. Host specificity of DNA produced by Escherichia coli. I. Host controlled modification of bacteriophage lambda [J]. J Mol Biol, 1962, 5: 18-36.
[9]DUSSOIX D, ARBER W. Host specificity of DNA produced by Escherichia coli. II. Control over acceptance of DNA from infecting phage lambda [J]. J Mol Biol, 1962, 5: 37-49.
[10]SMITH H O, WILCOX K W. A restriction enzyme from Hemophilus inf l uenzae. I. Purif i cation and general properties [J]. J Mol Biol, 1970, 51(2): 379-391.
[11]DANNA K, NATHANS D. Specif i c cleavage of simian virus 40 DNA by restriction endonuclease of Hemophilus influenza [J]. Proc Natl Acad Sci USA, 1971, 68(12): 2913- 2917.
[12]GREIDER C W, BLACKBURN E H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts [J]. Cell, 1985, 43(2): 405-413.
[13]JACKSON D A, SYMONS R H, BERG P. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli [J]. Proc Natl Acad Sci USA, 1972, 69(10): 2904-2909.
[14]SANGER F, COULSON A R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase [J]. J Mol Biol, 1975, 94(3): 441-448.
[15]SANGER F, NICKLEN S, COULSON A R. DNA sequencing with chain-terminating inhibitors [J]. Proc Natl Acad Sci USA, 1977, 74(12): 5463-5467.
[16]SAIKI R K, SCHARF S, FALOONA F, et al. Enzymatic amplif i cation of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia [J]. Science, 1985, 230(4732): 1350-1354.
(编辑:沈美芳)
Enzymes and life sciences (Ⅲ): Discovery and utilization of molecular biology enzymes
GUO Xiaoqiang
Department of Chemical Engineering, Shijiazhuang Vocational Technology Institute, Shijiazhuang 050081, China
In the last half of 20th century, molecular biology has made rapid development in life science. The discovery and application of molecular biology enzymes play an important role in promoting it. Several enzymes were identified and clarified including DNA polymerase, RNA polymerase, reverse transcriptase, restriction endonuclease, and telomerase. The progresses expand the understanding of many life phenomena. These enzymes also were used in molecular biology techniques, known as recombinant DNA, Sanger sequencing and polymerase chain reaction, which have a variety of applications such as nucleic acid manipulation, DNA sequencing and amplif i cation. In the paper, the history of researches on molecular biology enzymes was described to understand the signif i cance of these enzymes.
molecular biology enzyme, DNA polymerase, reverse transcriptase, restriction endonuclease, molecular biology technique, Nobel Prize
10.3969/j.issn.0253-9608.2015.05.008
†通信作者,E-mail:xiaoqiangguo123@163.com
猜你喜欢
中国生物化学与分子生物学报(2022年4期)2022-11-19
昆明医科大学学报(2022年2期)2022-03-29
中学生数理化·中考版(2021年9期)2021-11-20
植物保护(2021年4期)2021-11-12
生命科学研究(2021年5期)2021-11-11
科学24小时(2020年4期)2020-05-14
中国当代医药(2015年30期)2015-03-01
云南中医学院学报(2011年3期)2011-07-31
中学生天地·高中学习版(2008年1期)2008-02-19
中学英语之友·下(综合版)(2008年10期)2008-02-16
