
PPARα/PGC-1α在阿霉素扩张型心肌病小鼠心肌组织中的表达及作用*
2015-05-16 00:50王学胜杨永曜杨天和蒋清安铜仁市人民医院心内科贵州铜仁554300贵州省人民医院心内科贵州贵阳55000
中国病理生理杂志 2015年7期
王学胜,杨永曜,杨天和,蒋清安(铜仁市人民医院心内科,贵州铜仁554300;贵州省人民医院心内科,贵州贵阳55000)
PPARα/PGC-1α在阿霉素扩张型心肌病小鼠心肌组织中的表达及作用*
王学胜1,杨永曜2△,杨天和2,蒋清安2
(1铜仁市人民医院心内科,贵州铜仁554300;2贵州省人民医院心内科,贵州贵阳550002)
[摘要]目的:探讨过氧化物酶体增殖物激活受体α( peroxisome proliferator-activated receptors α,PPARα)及过氧化物酶体增殖物活化受体协同刺激因子1α( peroxisome proliferator activated receptor coactivator 1 alpha,PGC-1α)在阿霉素( doxorubicin,DOX)诱导扩张型心肌病小鼠心肌中的变化及其对心肌能量代谢及心功能的影响。方法:选取小鼠40只,分为正常对照组、单纯阿霉素模型组、PPARα抑制剂组和PPARα激动剂组。除正常对照组外,其余小鼠采用阿霉素诱导建立扩张型心肌病模型,检测其中PPARα及PGC-1α蛋白的表达,PPARα抑制剂组和PPARα激动剂组在造模前分别采用PPARα抑制剂及激动剂对小鼠预处理2周,高效液相层析法( high-performance liquid chromatography,HPLC)测量线粒体内腺苷酸含量,[3H]-ADP(氚标记二磷酸腺苷)掺入法检测线粒体膜腺苷酸转运体( adenine nucleotide translocator,ANT)转运活性,并利用超声心动图检测各组心脏结构及心功能。结果:成功建立阿霉素诱导的扩张型心肌病模型,PPARα及PGC-1α蛋白在模型组中表达明显低于正常组( P<0. 05),线粒体内高能磷酸盐含量和线粒体转运活性明显降低( P<0. 05),心功能显著下降,血流动力学指标紊乱( P<0. 05) ;与模型组比较,PPARα抑制剂预处理2周后,可显著降低PPARα/PGC-1α表达,线粒体内高能磷酸盐含量及线粒体ANT转运活性显著降低( P<0. 05),心功能恶化;而予PPARα激动剂预处理干预2周,上调PPARα/ PGC-1α蛋白表达同时,虽然线粒体内高能磷酸盐含量未发现有显著变化,但其可改善阿霉素心肌病大鼠线粒体ANT转运活性,超声心动图显示对心腔大小及心功能有改善作用( P<0. 05)。结论:在阿霉素诱导扩张性心肌病中,PPARα/PGC-1α对ANT的活性起重要调控作用,激活PPARα/PGC-1α可减轻阿霉素心肌损伤。
[关键词]过氧化物酶体增殖物活化受体α;过氧化物酶体增殖物活化受体协同刺激因子1α;阿霉素;扩张性心肌病;能量代谢
阿霉素( doxorubicin,DOX)在临床上广泛应用于治疗多种恶性肿瘤,但是因对心肌的毒性作用,大大限制了这一抗肿瘤药物的应用,随其用量的增加而引起的慢性心脏毒性会导致不可逆的心肌损伤,最终导致扩张性心肌病( dilated cardiomyopathy,DCM)及充血性心力衰竭[1]。DCM是国际医学界公认的难题[2]。目前对扩心病中心肌能量代谢变化及调控知之甚少,尤其是过氧化物酶体增殖物激活受体α ( peroxisome proliferator-activated receptors α,PPARα)及过氧化物酶体增殖物活化受体协同刺激因子1α ( peroxisome proliferator-activated receptor coactivator 1 alpha,PGC-1α)对药物性心肌病中能量代谢的转录调控鲜见报道。本研究拟通过阿霉素诱导建立小鼠扩张型心肌病模型,检测小鼠中PPARα及PGC-1α蛋白的表达,探讨其对阿霉素心肌病心力衰竭进程中心肌能量代谢及心衰进程的影响。
材料和方法
1实验动物
实验动物为FVB/NJ小鼠,共40只,鼠龄为10 ~12周,均由第三军医大学实验动物中心提供,生产合格证号为SCXX(军) 2002-007。
2主要试剂
PPARα抑制剂GW 6471购自天津彬馨博澳科技发展有限公司; PPARα特异性激活剂Wy 14643购自上海浩然生物技术有限公司;兔抗小鼠PPARα抗体购自Abcam;兔抗小鼠PGC-1α抗体购自CST;其它试剂为化学分析纯。
3实验方法
3.1动物分组及模型制备根据参考文献[3]改良制作阿霉素扩张型心肌病小鼠模型。模型组小鼠腹腔注射生理盐水溶解的阿霉素( 2. 0 mg·kg-1· d-1) 2周,间歇2周后再注射3周共7周,经动态心脏超声监测、静脉血B型脑钠肽( B-type natriuretic peptide,BNP)测定,与正常小鼠作对照,证实制造阿霉素慢性诱导的扩张型心肌病充血性心力衰竭模型成功。在模型建立前,将40只小鼠分为4组:正常对照( control)组、DOX扩心病模型组( DOX组)、PPARα抑制组( PPARα-组)、PPARα激活组( PPARα+组),每组10只。其中PPARα抑制组采用PPARα抑制剂GW 6471,PPARα激活组采用PPARα激动剂Wy14643。正常对照组小鼠腹腔注射与建模小鼠相同容量( 0. 1 mL)的生理盐水,PPARα抑制组予PPARα抑制剂GW 6471腹腔注射给药,给药浓度为20 mg·kg-1·d-1,PPARα激活组予PPARα特异性激活剂Wy 14643腹腔注射给药,给药浓度为20 mg·kg-1·d-1,时间均为2周。
3.2超声心动图检查剔除左胸前壁鼠毛,在Summit生产的台式麻醉机平台上,用4%异氟烷(以氧气为输送载体)进行吸入诱导麻醉,在小鼠进入外科深麻醉期后,以2%的浓度维持麻醉;使用GE心脏超声仪,将小动物15 MHz高频心脏超声探头放置于左胸前壁,切取满意的左室长轴切面后,在乳头肌水平将M型取样线垂直于左室后壁而获得M型超声心动图,随即获取主动脉流速曲线。选取3个连续心动周期,采用美国超声心动图协会推荐方法进行测量,测量数据包括左室舒张末期内径( end diastolic dimension,EDD)、左室收缩末期内径( end systolic diameter,ESD)、心率( heart rate,HR)、左室舒张末期容积( end diastolic volume,EDV)、左室收缩末期容积( end systolic volume,ESV),左室收缩功能由左室短轴缩短率( fractional shortening,FS)和左室射血分数( ejection fraction,EF)。HR由主动脉多普勒血流信号测量。FS、EDV、ESV及EF由以下公式计算: FS = ( EDD-ESD) /EDD; EDV =7×EDD3/( 2. 4 + EDD) ; ESV =7×ESD3/( 2. 4 + ESD) ; EF = ( EDV-ESV) / EDV×100%。
3.3血流动力学参数检测及留取心肌标本观察期满后,分别将各组大鼠用1%戊巴比妥钠( 30 mg/ kg)腹腔注射麻醉后,分离右颈动脉插管,应用Powerlab/8sp型8道生理记录仪( Adinstruments Pty)测量心率、平均动脉压( mean artery pressure,MAP)、主动脉收缩压( systolic aortic pressure,SAP)、主动脉舒张压( diastolic aortic pressure,DAP)、左室收缩压( left ventricular systolic pressure,LVSP)、左室舒张末压( left ventricular end diastolic pressure,LVEDP)和左室压力上升或下降最大速度(±dp/dtmax)。完成压力曲线记录后立即开胸取出心脏,用预冷的生理盐水灌注冲洗至冲洗液无红色。取离梗死处4 mm范围心室心肌冻存于液氮中,用于Western blot及ATP含量测定,其余部分用于线粒体分离。全过程于冰浴中进行。
3.4Western blot法检测PPARα及PGC-1α蛋白的表达取各组小鼠的心肌组织,研碎,提取总蛋白,蛋白定量后,取60 μg/well上样,行SDS-PAGE,转膜,分别与相关蛋白I抗孵育,再加对应的II抗孵育,ECL发光液显色,暗室压片,采集图像分析。
3.5心肌组织腺嘌呤核苷酸的测定参照Botker等[4]方法进行,色谱条件: Gilson系列高效液相色谱系统,UV检测器,检测波长254 nm,灵敏度0. 01 AUFS。色谱柱为4. 6 nm×250 nm,YWG-ODS C1810 μm;流动相为2 mmol/L PBS( pH = 5. 5),样本加入量20 μL。采用衡速洗脱,全程20 min,流速1 mL/ min。
3.6心肌线粒体ANT转运活性的测定用抑制剂终止法测定ANT转运活性。取制备好的线粒体悬液50 μL分离介质稀释后加入0. 3 μmol/L的[3H]-ADP溶液20 μL,10 s,立即加入3. 2 nmol/L ANT抑制剂ATR 50 μL,迅速混匀终止ANT转运功能。4℃、12 000 r/min离心20 min,弃上清,沉淀以1 mL分离介质溶解清洗后加入8. 8 mol/L H2O2400 μL,置于70℃水浴消化40 min,取200 μL用液体闪烁计数法测放射性活性。对照管在加入[3H]-ADP前先加入苍术苷,用实验管的放射性活性减去对照管放射性活性即为ANT的转运活性,结果以ADP含量( nmol·min-1·g-1)表示。
4统计学处理
实验所有数据均采用SPSS 17. 0进行计算。计量资料采用均数±标准差( mean±SD)表示,组间比较采用单因素方差分析,组间两两比较采用LSD法计算。以P<0. 05为差异有统计学意义。
结果
1阿霉素诱导扩张性心肌病模型的建立
通过对FVB/NJ小鼠行DOX腹腔注射慢性中毒诱导( 2 mg·kg-1·d-1),用药7周,中间间歇2周,DOX总剂量( 20 mg/kg),我们成功建立FVB/NJ小鼠扩张型心肌病充血性心力衰竭模型,单纯DOX扩心病模型组死亡率仅2%,各项观察指标包括超声心动图检查、左心室内径测量、心肌病理切片结果等均符合扩张型心肌病的改变;经统计学处理,对比正常组可见,模型组第9周左室EDD、ESD及BNP显著增加,IVS、PWT、FS及EF明显下降,PPARα抑制组死亡率40%,而PPARα激活组死亡率10%,见图1、表1。

Figure 1.The results of echocardiography and myocardial HE staining in DCM and control group.图1 阿霉素扩张型心肌病心衰模型超声心动图及HE染色图片
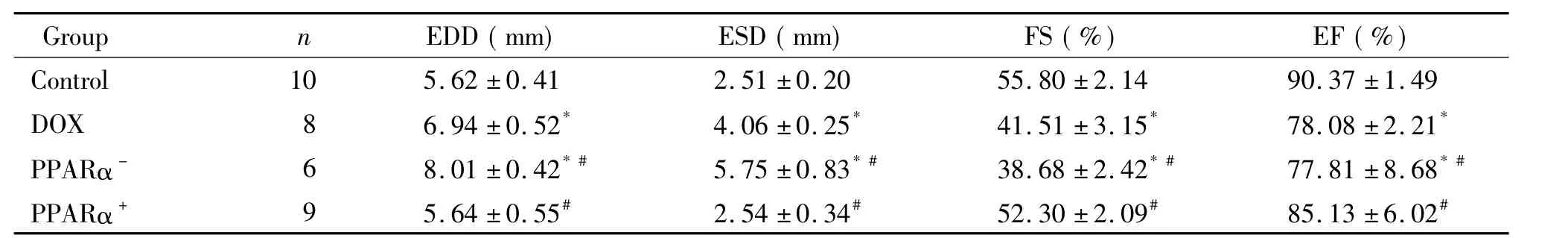
表1 各组小鼠心脏超声检查结果Table 1.Echocardiographic results of the mice in different groups ( Mean±SD)
2各组大鼠心肌PPARα及PGC-1α的蛋白表达变化
与正常组比较,DOX模型组、给予PPARα抑制剂组及给予PPARα激动剂组PPARα及PGC-1α蛋白表达明显下降;而相对DOX模型组,PPARα+组的PPARα和PGC-1α蛋白表达明显上调,PPARα-组PPARα和PGC-1α蛋白明显下调,差异显著( P<0. 05),见图2。

Figure 2.The protein expression of PPARα and PGC-1α in the myocardium determined by Western blot.Mean±SD.n =4.*P<0. 05 vs control group;#P<0. 05 vs DOX group.图2 各组小鼠心肌PPARα及PGC-1α蛋白的表达
3心脏超声检查结果
与正常对照组相比,单纯DOX模型组左室EDD 及ESD明显升高( P<0. 05),FS及EF均显著降低( P<0. 05),PPARα+组与正常对照组比较,虽然EDD、ESD值较大,EF、FS有所下降,但未见明显统计学差异。经统计,PPARα+组EDD、ESD较DOX组显著缩小,而FS、EF较DOX组明显著升高( P<0. 05),PPARα-组心脏超声检查显示EDD和ESD明显高于正常对照组,FS及EF明显低于对照组( P<0. 05),见图3、表1。
4血流动力学检测结果
与正常对照组比较,DOX模型组LVSP、+ dp/ dtmax及-dp/dtmax水平显著降低( P<0. 05),而LVEDP较正常对照组高,差异显著( P<0. 05)。PPARα+组与正常对照组各血流动力学检查未见明显统计学差异,PPARα-组血流动力学检查显示LVSP、+ dp/dtmax及-dp/dtmax水平明显低于正常对照组,而LVEDP水平明显高于对照组( P<0. 05),见表2。
5各组小鼠心肌线粒体内腺苷酸含量变化
与正常组相比,DOX组、PPARα+组及PPARα-组小鼠心肌线粒体内高能磷酸盐三磷酸腺苷( adenosine-5’-triphosphoric acid,ATP)、二磷酸腺苷( adenosine-5’-diphosphoric acid,ADP)与一磷酸腺苷( adenosine-5’-monophosphate,AMP)均显著降低( P<0. 05) ;与DOX组相比,PPARα+组大鼠心肌线粒体内高能磷酸盐含量虽然有增高趋势,但差异不显著,见图4。
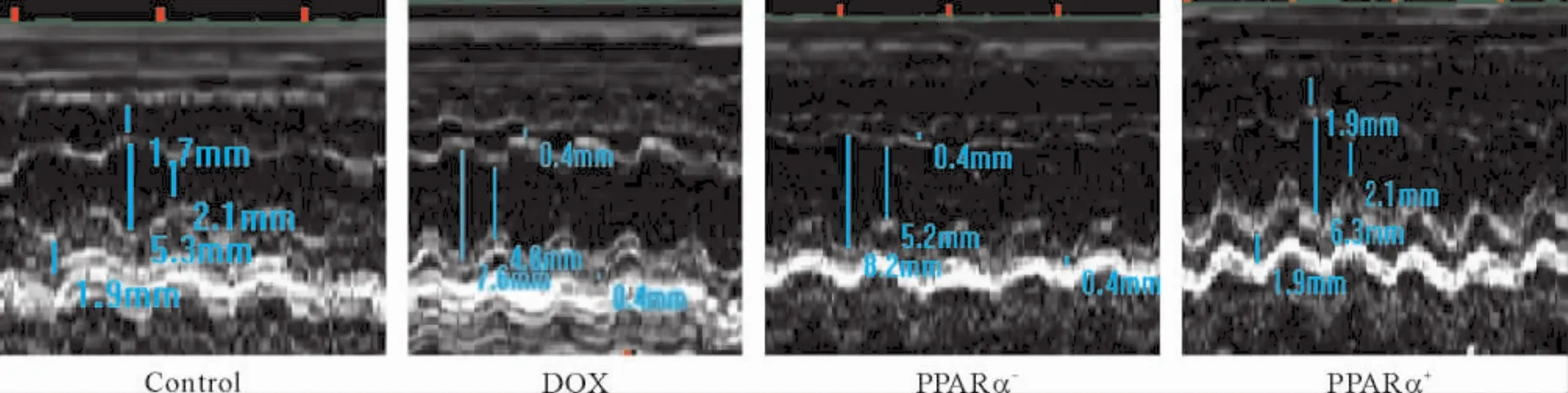
Figure 3.The results of echocardiography in the mice with different treatments.图3 各组小鼠典型心脏超声影像

表2 各组小鼠血流动力学检测结果Table 2.Hemodynamics results of the mice in different groups( Mean±SD)
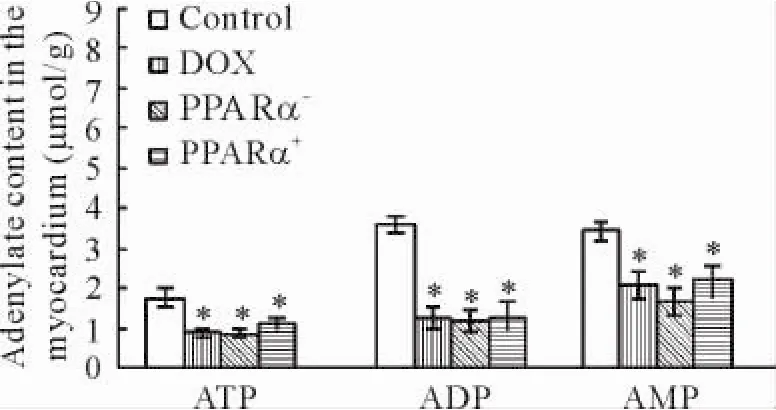
Figure 4.The changes of adenylate content in the myocardium in different groups.ATP: adenosine-5’-triphosphoric acid; ADP: adenosine-5’-diphosphoric acid; AMP: adenosine-5’-monophosphate.Mean±SD.n = 5.*P<0. 05 vs control group.图4 大鼠心肌线粒体内高能磷酸盐含量的改变
6各组小鼠心肌线粒体ANT转运活性的变化
与正常组相比,阿霉素心肌病小鼠的心肌ANT转运活性均降低( P<0. 05) ;而PPARα-组心肌ANT转运活性相对DOX组更为降低( P<0. 05),见图5。
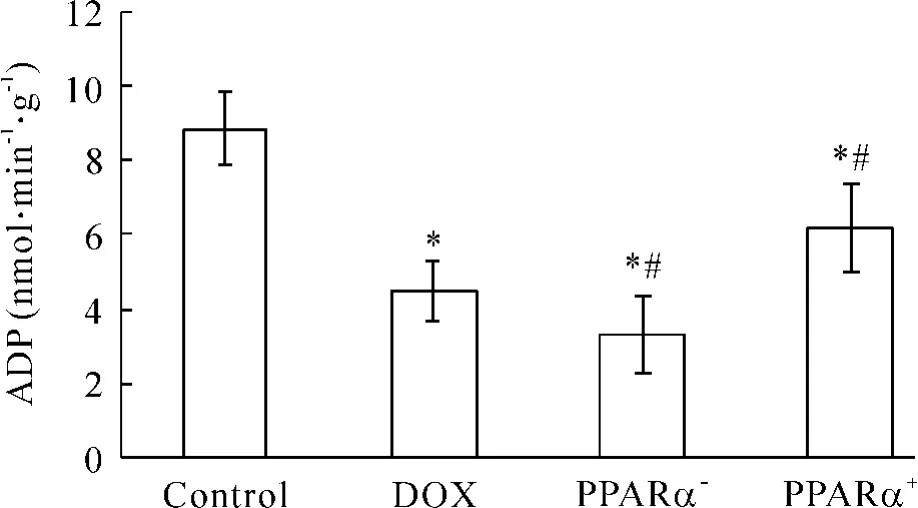
Figure 5.The changes of myocardial mitochondrial ANT transport activity in different groups.Mean±SD.n = 5.*P<0. 05 vs control group;#P<0. 05 vs DOX group.图5 各组大鼠心肌线粒体ANT转运活性的比较
讨论
阿霉素为临床常用的广谱、高效蒽环类抗肿瘤药,广泛用于急慢性白血病、各种实体瘤等多种癌症的化疗,效果显著。然而,DOX明显的、不可逆的剂量依赖性心脏毒性,严重限制其临床应用,其由于取材方便,方法简单,结果满意,其常被用于诱导非缺血性心肌病模型。目前国内、外多采用兔、大鼠作为阿霉素的诱导对象,但采用这2种动物为对象具有体重、大、用药多、费用高、死亡率高的缺点[5-6]。为此,本研究采用FVB/NJ小鼠作为诱导对象,以DOX腹腔注射慢性中毒诱导( 2 mg·kg-1·d-1),用药7周,中间间歇2周,DOX总剂量( 20 mg/kg),用药规律基本符合阿霉素临床用药规律,成功建立FVB/NJ小鼠扩张型心肌病充血性心力衰竭模型,模型成功率为98%,各项观察指标包括超声心动图检查、左心室内径测量、心肌病理切片结果等均符合扩张型心肌病的改变[7],具有经济、可靠、符合临床用药疗程等优点,为阿霉素心肌病研究的顺利进行提供了保障。
目前,阿霉素心肌病的发病机制仍未阐明。一般认为,在DOX引起的心脏毒性发病过程中,线粒体是主要靶向器官,DOX亲和心肌后,阿霉素氧化还原为半醌,产生大量氧自由基直接攻击线粒体,发生线粒体结构和功能障碍,表现为线粒体肿胀、裂解、嵴断裂直至溶解,使其氧化磷酸化受到抑制,ATP生成障碍,严重影响心肌的收缩和舒张功能[8-9]。
过氧化物酶体增生物激活受体( peroxisome proliferator-activated receptors,PPARs)是心肌脂质和能量代谢的重要调控子,调控出生后心肌线粒体编码的脂肪酸β氧化酶的大部分核基因表达[10]。PGC-1α是PGC-1家族被发现的第1个成员,随后PGC-1α的同源蛋白PGC-1β以及PRC陆续被发现,构成一个小的辅激活因子家族,PGC-1是线粒体生物合成和呼吸的一个关键调节点[11-13]。有人研究发现通过上调PGC-1通路可以调节线粒体生物合成,从而预防和逆转各种疾病的战略,包括肥胖和糖尿病[14]。在对心肌缺血/再灌注、心肌梗死、压力负荷性心衰动物模型的研究中发现PPARα/PGC-1α对心功能产生了有益作用,但其在阿霉素诱导的心肌病中的作用尚不明确。
本研究采用上述阿霉素诱导的小鼠扩张型心肌病模型,检测其中PPARα及PGC-1α蛋白的表达,并在造模成功后分别采用PPARα抑制剂及激动剂对模型进行处理,检测各组超声心动图和血流动力学变化情况。结果显示PPARα及PGC-1α蛋白在空白模型组、PPARα抑制剂及激动剂组中表达明显低于正常组。超声心动图显示DOX诱导组左室内径增大,心功能降低,采用PPARα抑制剂处理的小鼠其左室舒张末期或收缩末期内径、左室短轴缩短率及左心室射血分数均更加恶化,而采用PPARα激动剂组的小鼠其上述指标较DOX诱导组及PPARα抑制剂组改善。进一步地,我们对各组小鼠的血流动力学指标也进行了检测,其结果显示模型组及PPARα抑制剂组左心室收缩压、收缩期左室发展压最大变化速率、舒张末期左室发展压最大变化速率均较正常组别有明显的降低,而左室舒张末压则相对较高,而PPARα激动剂组血流动力学紊乱情况明显改善,和心脏超声检测结果相符。
同时本研究发现,在阿霉素诱导扩张型心肌病后,通过激活PPARα/PGC-1α,升高PPARα/PGC-1α蛋白表达的同时,虽然线粒体内高能磷酸盐含量未发现有显著变化,但明显改善阿霉素心肌病小鼠线粒体ANT转运活性,对血流动力学指标有改善作用,延缓了心衰的发展,而予抑制PPARα/PGC-1α时则产生相反的作用,促进心腔扩张和心衰的进展。既往研究发现通过苯扎贝特药理过程或者PPARα-PGC-1α转基因过表达途径代谢调节产生的线粒体生物合成对小鼠线粒体肌病模型有显著效果,认为PPARα-PGC-1α途径将成为治疗线粒体肌病的一种有效手段[15]。本研究从正反两方面干预PPARα-PGC-1α途径,更进一步证实,针对线粒体保护的治疗方法对DOX诱导的心肌病是有效的,其机制可能与通过保护线粒体、上调凋亡调控基因的表达抑制阿霉素诱导的心肌细胞凋亡等有关。
综上,本实验揭示了阿霉素诱导的扩张型心肌病小鼠中PPARα及PGC-1α呈低表达,从正反两方面观察平衡调控心肌能量代谢对阿霉素诱导的充血性心力衰竭的影响,证实促进PPARα/PGC-1α的激活,可减轻阿霉素诱导的心肌损伤,为今后该类疾病的防治提供了新的潜在治疗靶点,其具体机制值得进一步研究。
[参考文献]
[1]Durante-Mangoni E,Maiello C,Limongelli G,et al.Management of immunosuppression and antiviral treatment before and after heart transplant for HIV-associated dilated cardiomyopathy[J].Int J Immunopathol Pharmacol,2014,27( 1) : 113-120.
[2]Sbrana F,Greco P,Rovai D.Dilated cardiomyopathy postchemotherapy[J].Recenti Prog Med,2009,100( 3) : 132-136.
[3]周岩,赵丽蓉,杨思睿.阿霉素诱导大鼠扩张型心肌病模型的建立[J].中国实验诊断学,2010,14( 3) : 321-334.
[4]Botker HE,Kimose HH,Helligso P,et al.Analytical evaluation of high energy phosphate determination by high performance liquid chromatography in myocardial tissue [J].J Mol Cell Cardiol,1994,26( 1) : 41-48.
[5]Gava FN,Zacché E,Ortiz EM,et al.Doxorubicin induced dilated cardiomyopathy in a rabbit model: an update [J].Res Vet Sci,2013,94( 1) : 115-121.
[6]黄荣杰,刘唐威,伍伟锋,等.呋喃唑酮制备大鼠扩张型心肌病模型[J].中国病理生理杂志,2005,21 ( 11) : 2147-2150.
[7]曹文军,李良,刘国贞,等.扩张性心肌病的病理形态学观察[J].中国病理生理杂志,2001,17( 8) : 755-758.
[8]Nithipongvanitch R,Ittarat W,Cole MP,et al.Mitochondrial and nuclear p53 localization in cardiomyocytes: redox modulation by doxorubicin ( adriamycin) ?[J].Antioxid Redox Signal,2007,9( 7) : 1001-1008.
[9]Yang F,Teves SS,Kemp CJ,et al.Doxorubicin,DNA torsion,and chromatin dynamics[J].Biochim Biophys Acta,2014,1845( 1) : 84-89.
[10]Dirks-Naylor AJ.The role of autophagy in doxorubicin-induced cardiotoxicity[J].Life Sci,2013,93( 24) : 913-914.
[11]Domenici FA,Brochado MJ,Martinelli AL,et al.Peroxisome proliferator-activated receptors alpha and gamma2 polymorphisms in nonalcoholic fatty liver disease: a study in Brazilian patients[J].Gene,2013,529( 2) : 326-331.
[12]Gleyzer N,Scarpulla RC.Activation of a PGC-1-related coactivator ( PRC) -dependent inflammatory stress program linked to apoptosis and premature senescence[J].J Biol Chem,2013,288( 12) : 8004-8015.
[13]Scarpulla RC.Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator[J].Ann NY Acad Sci,2008,1147: 321-334.
[14]Lu Z,Xu X,Hu X,et al.PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload[J].Antioxid Redox Signal,2010,13( 7) : 1011-1022.
[15]Wenz T,Diaz F,Spiegelman BM,et al.Activation of the PPAR/PGC-1α pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype [J].Cell Metab,2008,8( 3) : 249-256.
Role of PPARα/PGC-1α in doxorubicin induced mouse dilated cardiomyopathy
WANG Xue-sheng1,YANG Yong-yao2,YANG Tian-he2,JIANG Qing-an2
(1Department of Cardiology,The People’s Hospital of Tongren,Tongren 554300,China;2Department of Cardiology,Guizhou Provincial People’s Hospital,Guiyang 550002,China.E-mail: yangyy19@ hotmail.com)
[ABSTRACT]AIM: To investigate the changes of peroxisome proliferator-activated receptors ( PPAR)α/peroxisome proliferator activated receptor coactivator 1 alpha ( PGC-1α) in doxorubicin ( DOX) induced dilated cardiomyopathy ( DCM) and its effect on the energy metabolism and myocardial function in mice.METHODS: Forty mice were randomly divided into 4 groups: control group,DOX group,PPARα inhibitor group and PPARα agonist group.The DCM model was established by injection of DOX.The protein levels of PPARα/PGC-1α were detected.The PPARα inhibitor and PPARα agonist were used 2 weeks beforeinjection of DOX.The contents of adenine acid and phosphocreatine ( Pcr) in the mitochondria were measured by high-performance liquid chromatography ( HPLC).The ANT activity was analyzed by the atractyloside-inhibitor stop technique.The changes of the echocardiography and hemodynamics were also observed.RESULTS: DOX induced DCM model was successfully established.The protein levels of PPARα and PGC-1α in control group were significantly higher than those in DOX group ( P<0. 05).Both of the high-energy phosphate contents and the transport activity of ANT were decreased in DOX group ( P<0. 05),and the hemodynamic parameters were disordered ( P<0. 01).Compared with DOX group,PPARα inhibitor pre-treatment significantly reduced the PPARα/PGC-1α expression.Mean-book=1161,ebook=17while,high-energy phosphate contents in the mitochondria and the ANT transport activity of the mitochondria decreased,as well as the left ventricular function ( P<0. 05).On the other hand,PPARα agonist significantly increased the expression of PPARα and PGC-1α,and improved the transport activity of ANT.In addition,the hemodynamic parameters were ameliorated,but the high-energy phosphate contents of the mitochondria did not significantly change.CONCLUSION: PPARα/PGC-1α plays an important role in the regulation of ANT transport activity in dilated cardiomyopathy induced by DOX,and the activation of PPARα/PGC-1α has protective effects on the DCM induced by DOX.
[KEY WORDS]Peroxisome proliferator-activated receptors α; Peroxisome proliferator activated receptor coactivator 1α; Doxorubicin; Dilated cardiomyopathy; Energy metabolism
通讯作者△Tel: 0851-5937213; E-mail: yangyy19@ hotmail.com
*[基金项目]国家自然科学基金资助项目( No.81260053)
[收稿日期]2015-01-04[修回日期]2015-02-11
[文章编号]1000-4718( 2015)07-1160-06
[中图分类号]R541. 6; R363.2
[文献标志码]A
doi:10.3969/j.issn.1000-4718.2015.07.002
猜你喜欢
临床军医杂志(2022年8期)2022-08-25
世界科学技术-中医药现代化(2022年2期)2022-05-25
中国典型病例大全(2022年11期)2022-05-13
成都信息工程大学学报(2021年6期)2021-02-12
天津医科大学学报(2019年6期)2019-08-13
肿瘤防治研究(2019年7期)2019-08-01
中国临床医学影像杂志(2019年1期)2019-04-25
中国计划生育学杂志(2017年3期)2017-06-01
中国当代医药(2015年31期)2015-03-01
郑州大学学报(医学版)(2015年1期)2015-02-27
