
Improved dissolution and bioavailability of silymarin delivered by a solid dispersion prepared using supercritical f l uids
2015-05-15 13:09:04GngYngYpingZhoNinpingFengYongtiZhngYingLiuBeileiDng
Gng Yng,Yping Zho,Ninping Feng,*,Yongti Zhng, Ying Liu,Beilei Dng
aSchool of Pharmacy,Shanghai University of Traditional Chinese Medicine,Shanghai 201203,China
bSchool of Chemistry and Chemical Engineering,Shanghai Jiao Tong University,Shanghai 200240,China
Original Research Paper
Improved dissolution and bioavailability of silymarin delivered by a solid dispersion prepared using supercritical f l uids
Gang Yanga,Yaping Zhaob,Nianping Fenga,*,Yongtai Zhanga, Ying Liua,Beilei Danga
aSchool of Pharmacy,Shanghai University of Traditional Chinese Medicine,Shanghai 201203,China
bSchool of Chemistry and Chemical Engineering,Shanghai Jiao Tong University,Shanghai 200240,China
ARTICLEINFO
Article history:
Received 8 October 2014
Received in revised form
8 December 2014
Accepted 13 December 2014
Available online 26 December 2014
Silymarin
Solution-enhanced dispersion by
supercritical f l uids
Solid dispersion
Dissolution
Bioavailability
The objective of this study was to improve the dissolution and bioavailability of silymarin (SM).Solid dispersions(SDs)were prepared using solution-enhanced dispersion by supercritical f l uids(SEDS)and evaluated in vitro and in vivo,compared with pure SM powder. The particle sizes,stability,and contents of residual solvent of the prepared SM-SDs with SEDS and solvent evaporation(SE)were investigated.Four polymer matrix materials were evaluated for the preparation of SM-SD-SEDS,and the hydrophilic polymer,polyvinyl pyrrolidone K17,was selected with a ratio of 1:5 between SM and the polymer.Physicochemical analyses using X-ray diffraction and differential scanning calorimetry indicated that SM was dispersed in SD in an amorphous state.The optimized SM-SD-SEDS showed no loss of SM after storage for 6 months and negligible residual solvent(ethanol)was detected using gas chromatography.In vitro drug release was increased from the SM-SDSEDS,as compared with pure SM powder or SM-SD-SE.In vivo,the area under the rat plasma SM concentration-time curve and the maximum plasma SM concentration were 2.4-fold and 1.9-fold higher,respectively,after oral administration of SM-SD-SEDS as compared with an aqueous SM suspension.These results illustrated the potential of using SEDS to prepare SM-SD,further improving the biopharmaceutical properties of this compound.
©2014 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/ licenses/by-nc-nd/4.0/).
1.Introduction
Silymarin(SM),an extract of Silybum marianum(L.),contains a mixture of four fl avonolignan isomers:silibinin(70-80%), silycristin(20%),silydianin(10%),and isosilybin(0.5%)[1,2]. Silybin is therefore the major component of SM and is responsible for its pharmacological activity.SM has traditionally been self-administered for the treatment of liver disorders[3,4].The effects of SM on the liver have been attributed to its inhibition of hepatotoxin binding to receptor sites on the hepatocyte membrane;inhibition of glutathione oxidation,increasing its levels in the liver and intestine; antioxidant activity;and stimulation of ribosomal RNA polymeraseandsubsequentproteinsynthesis,leadingto enhanced hepatocyte regeneration[5].However,the effectiveness of SM as a liver disease remedy is limited by its poor aqueous solubility,resulting in low oral bioavailability due to poor enteral absorption[6].Recently,various vehicles have been employed to improve the solubility and bioavailability of SM,such as solid lipid nanoparticles,microemulsion systems, liposomes,and solid dispersions(SDs)[7-10].
SDs have been used widely to enhance the dissolution rate of drugs with low aqueous solubility.In SD systems,a drug may exist as an amorphous form within a polymeric carrier. This may result in an increased drug dissolution rate,as compared with its crystalline form.The mechanisms involved in this SD-mediated enhancement of drug dissolution have been proposed by several investigators[11,12].
SDs can be prepared using a range of methods,such as melting,solvent evaporation,solvent melting,spray-drying, and supercritical fl uid techniques.Supercritical fl uid approaches have several advantages over more conventional preparation methods,including the ability to reduce residual organic solvent levels and to produce SDs with smaller particle sizes and better fl owability[13].There are several variants of supercritical fl uid techniques,including rapid expansion of supercriticalsolutions,particlesfromgas-saturatedsolutions,gas antisolvent process,supercritical anti-solvent process,precipitationfromcompressedanti-solvent,aerosolsolventextraction system,and solution-enhanced dispersion by supercritical fl uids(SEDS).SEDSprovidesthemostpromisingmethodforthe preparation of SD[14].It uses semi-continuous processes to atomize the solution into a supercritical atmosphere.Provided thatthedrugissparinglysolubleinthesupercritical fl uid,which is highly soluble in the solvent,the supercritical fl uid in the solventdropletscanproduceanantisolventeffect.Thisprocess producessuper-saturatedsolutions,facilitatingprecipitationof the solid in the form of small particles[15].
In the current study,SEDS was used to establish an SD delivery system for oral administration of SM,with the aim of enhancing drug dissolution and bioavailability.The physicochemical properties of SD prepared using several polymer materials were investigated,including polyvinyl pyrrolidone (PVP)K17,PVP K30,hydroxypropyl methylcellulose(HPMC) K4M,and HPMC K15M.In addition,analyses of stability and residual solvent were performed to compare SM-SD prepared by SEDS and by solvent evaporation(SE).In vitro dissolution and in vivo pharmacokinetics were analyzed to assess the SMSD-SEDS.
2.Materials and methods
2.1.Materials
SM(purity>80%)and PVP K17 were purchased from Dalian Meilun Biotech Co.,Ltd.(Dalian,China).PVP K30 was obtained from Sinopharm Chemical Reagent Co.,Ltd.(Shanghai, China).HPMC K4M and HPMC K15M were supplied from Colorcon(USA).Carbon dioxide(CO2)with a purity of 99.99%was obtained from Shanghai Jiao Tong University(Shanghai, China).All other chemicals were reagent grade and used as received.
2.2.Animals
Animal studies of male Wistar rats weighing 250±10 g were conducted with the approval of the Animal Ethical Committee,Shanghai University of Traditional Chinese Medicine.The animals were kept in an agreeable environment with free access to a rodent diet and water and were acclimatized for at least 1 week before the start of the study.
2.3.Preparation of SM-SD
The supercritical pilot plant at Nantong Huaxing Petroleum Devices Co.,Ltd.(Nantong,China),shown in a schematic diagram in Fig.1A,was employed in this study.Brief l y,this apparatus included three major components:a CO2delivery system,an organic solution delivery system,and a precipitation system.The supercritical CO2and the organic solution wereseparatelypumpedintothehigh pressurevesselthrough different inlets of the coaxial nozzle(the diameter of inner tubule was 0.2 mm and diameter of outside part was 1 mm) and continuously discharged from the bottom.The inner structure of the nozzle is shown in Fig.1B.For preparation of SM-SD,CO2from the cylinder(Fig.1A,1)was refrigerated(2) and compressed to 15 MPa by the high-pressure pump(3) before the temperature controlling system(6)was activated to increase the temperature to 50°C.The pressure of the precipitation system was increased by injection of CO2until the pressure reached 15 MPa.After the pressure and temperature of the view vessel(9)reached the required values,valve C was adjusted to maintain constant pressure in the vessel.Then SM and the excipients with the weight ratio of 1:5(based on the results of preliminary experiment)were separately dissolved or dispersed in ethanol and a mixture of dichloromethane and ethanol(3/2,v/v).The solution was aspirated by a high pressure constant f l ow pump(11)(LC100,Nantong,China)at a f l ow rate of 1 ml/min.Supercritical CO2and the organic solution mixed and diffused rapidly.Solutes originally dissolved in the organic solvent rapidly reached super-saturation,resulting in the precipitation of SM-SD-SEDS in the vessel.Once the solution was exhausted,valve B was closed,and supercritical CO2was continuously pumped for about 40 min in order to remove residual organic solvent from the SM-SD.After that, valve A was closed while valve C remained open.The pressure of the precipitation vessel was slowly reduced and the product in the vessel was collected for further use.
SM-SDs were also prepared using the SE method,whereby the drug and matrix were both dissolved in ethanol,and the solvent was removed under vacuum in a rotary evaporator(R-205,Shanghai,China).The resultant SM-SD-SE was placed in a vacuum drying oven(DHG-9070A,Shanghai,China)to harden prior to pulverization,sieving through a 250-μm sieve,and storage in a desiccator.
2.4.High-performance liquid chromatography(HPLC) analysis
The content of SM was determined using an HPLC system(LC-2010A HT,Shimadzu,Japan)with an Agilent Eclipse XDB-C18 column(5 μm,4.6×250 mm)(Agilent,Shanghai,China).The mobile phase consisted of methanol and pure water(46:54,v/ v)at a f l ow rate of 0.8 ml/min.The eff l uent was monitored at 288 nm.
2.5.In vitro dissolution
The rate of dissolution of SM from the indicated preparations was evaluated using the small vessel method.The dissolution medium was phosphate buffer solution(pH 6.8)containing 0.3%(w/v)sodiumdodecylsulfateandmaintainedat 37±0.5°C stirring at 100 rpm.Samples of 2.0 ml were withdrawn after 5,10,15,20,30,60,90,120 min,and replaced by the same volume of fresh medium at 37±0.5°C.The samples were f i ltered through a 0.45-μm membrane,and the SM contentwasdetermined by HPLC,as describedin Section2.4.Each experiment was carried out in triplicate.
2.6.Scanning electron microscopy(SEM)
The SD surface morphology was observed by SEM(NOVA NanoSEM 250,Shanghai,China).Samples were sprayed with gold before examination.Microphotographs were obtained using an accelerating voltage of 5 kV at 1000-25,000× magnif i cation.
2.7.X-ray diffraction(XRD)
SampleswereanalyzedusinganX-raypolycrystalline diffractometer(D8 ADVANCE,Bruker,Germany).Cu Kα radiation was used as the X-ray source.Samples were scanned over the range of 50°-70°2θ at a scanning speed of 6°/min and a step size of 0.02°.The voltage was set at 40 kV and the current was 40 mA.
2.8.Differential scanning calorimetry(DSC)
The thermal behaviors of the SD samples were analyzed using a differential scanning calorimeter(Q2000,New Castle,USA). SD(~5-10 mg)was sealed in a specialized aluminum pan using an aluminum lid with a pinhole.Measurements were performed over 40-250°C under a dry nitrogen atmosphere at a heating rate of 10°C/min.
2.9.Stability studies
The SM content of the SM-SD preparations was analyzed at 0, 1,2,4,and 6 months to compare the degradation over time, and XRD was used to investigate the physical stability of SM-SD.
2.10.Determination of residual solvent
The ethanol content of the SM-SD preparations was analyzed by gas chromatography to monitor the eff i ciency of solvent removal during SEDS.The samples were dissolved in acetone.An Agilent 7890A gas chromatography system coupled with a fl ame ionization detector(Agilent,California,USA)and an Agilent19091N-113HP-INNOWAXcapillarycolumn (30 m×0.32 mm×0.25 μm,Agilent,California,USA)was employed,using nitrogen as the carrier gas.The 20:1 split mode was used,while the injection port was operated at 150°C.The initial oven temperature was held at 50°C for 10 min,and then raised to 200°C for 10 min at a heating rate of 5°C/min.The temperature of hydrogen fl ame ionization detector was set at 250°C.
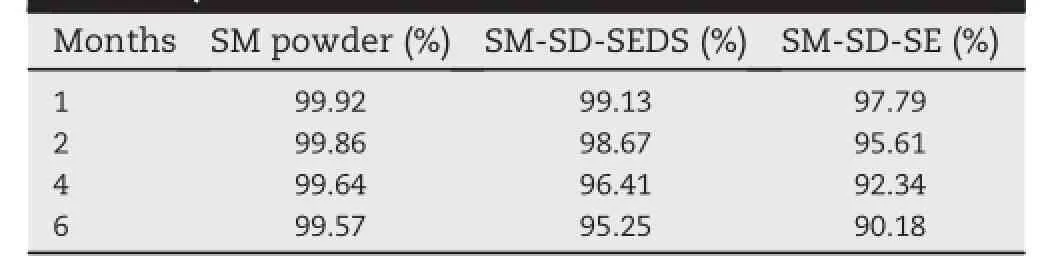
Table 1-The silymarin(SM)content of the indicated preparations after storage at room temperature for the indicated time periods(as a percentage of the level at 0 month).
2.11.In vivo pharmacokinetics
Male Wistar rats(Section 2.2)were randomly divided into two groups(n=6/group).The animals were starved for 12 h before the experiment but had free access to water.One group of rats was orally administered 20 mg/kg SM,contained withinSM-SD-SEDS,and the second group of rats received an aqueous suspension of SM(prepared by simply adding SM to purif i ed water).About 0.3-ml blood samples were collected from the eye socket vein and placed in heparinized tubes at the indicated time points after dosing.The blood samples were centrifuged(Eppendorf,Hamburg,Germany)at 5000 rpm for 10 min to isolate the plasma.One milliliter of ethyl acetate was added to the plasma and vortex-mixed(IKA,Staufen,Germany)for 1 min,and then centrifuged at 10,000 rpm for 10 min.The supernatant layer was dried under nitrogen and redissolved in 100 μl methanol prior to HPLC determination,as described in Section 2.4.
2.12.Statistical analysis
All the generated data were presented as mean±standard deviation.The pharmacokinetic parameters were estimated by a non-compartmental model using DAS 2.0 software.Statistical analyses were conducted by one-way analysis of variance(ANOVA).A value of P<0.05 was considered statistically signif i cant.
3.Results and discussion
3.1.In vitro SM release from SD prepared using various polymeric matrices
In this study,SM-SDs were successfully prepared by SEDS using each of the following excipients;PVP K17,PVP K30, HPMC K4M,and HPMC K15M.The cumulative release of SM from the formulated SDs(Fig.2)was markedly improved as compared with SM powder.There were signif i cant differences between the samples in terms of their in vitro SM release (ANOVA,P<0.05).The PVP K17-based SD showed the most effective SM release over the test period,achieving 98%cumulative drug release in 15 min.These results demonstrated that the SDs improved SM dissolution,as compared with the raw drug powder,in the following order:SM powder<HPMC K15M<HPMC K4M<PVP K30<PVP K17.As previously reported,PVP K17 forms reticulate structures and incorporation of drug molecules into these results in molecular dispersion [16],which may contribute to increased drug dissolution and release.
3.2.SEM
Fig.3 shows SEM images of SM powder,PVP K17,a physical mixture of SM and PVP K17,and SM-SD generated by SE or SEDS.The physical mixture of SM and PVP K17 had a different morphology to that of SM-SD-SE and SM-SD-SEDS,with the SM-SD-SEDS showing the smaller size distribution;this may contribute to enhanced drug solubility.
3.3.DSC
Sample DSC curves are shown in Fig.4.For SM powder,a sharp endothermic peak was observed at 169.98°C,corresponding to its melting point.The melting endotherm for PVP K17 was observed at 204.82°C.The DSC analysis showed no relocation of peaks in the physical mixture,where the characteristic melting points of PVP K17 and SM remained unchanged.This indicated that there were no chemical drug-polymer interactions.However,the endothermic peak of SM was not observed in SDs prepared by SEDS and SE,and the endothermic peak of PVP K17 had shifted.These results suggested that SM was present in a different state in the SM-SD.
3.4.XRD
To examine crystal morphology,XRD analyses were carried out and the patterns generated by SM,PVP K17,the physical mixture of PVP K17 and SM,and SM-SD prepared by SEDS and SE were presented in Fig.5.SM powder showed a crystalline structure,as demonstrated by its sharp and intense diffraction peaks.These diffraction peaks were retained in the physical mixture,suggesting that SM remainedin a crystalline state.In contrast,these characteristic peaks were absent in SM-SDs,indicating that SM was in an amorphous state.These XRD results were in agreement with the DSC results.The amorphous state of SM within SDs may contribute to increased drug solubility[17,18].
3.5.Stability
SDs have been demonstrated to be effective formulations for the improvement of drug solubility,but they may have a critical drawback in terms of stability.The recrystallization of amorphous drugs in SD may lead to a reduced dissolution rate and consequently a lower bioavailability[19,20].The data generated by SM content analysis and XRD of SM-SD were presented in Table 1 and Fig.6.The SM content of SM powder and SM-SD produced by SEDS or SE was not signif i cantly changed following storage for six months.However,more drugloss was observed in SM-SD-SEthanin SM-SD-SEDS.This may ref l ect increased molecular motion within SM-SD-SE, recrystallization of the amorphous drug,and the associated chemical degradation[21,22].Furthermore,the XRD result for SM-SD-SEDS was similar on day 0 and at 6 months(Fig.6), indicating that the drug remained in an amorphous state throughout the test period.However,the prof i le from SM-SDSE showed a sharp peak at 21°2θ,which could suggest a certain degree of drug recrystallization in this formulation. Previous reports have indicated that residual solvent in SDs could promote nucleation for crystallization and therebypromote degradation of the active ingredients.This may therefore have contributed to the greater drug loss and the altered XRD of SM-SD-SE,as compared with SM-SD-SEDS.
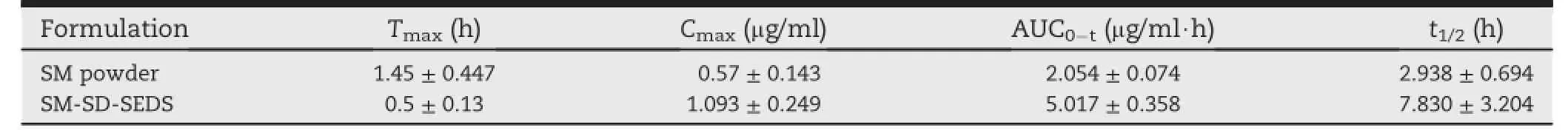
Table 2-6).The main pharmacokinetic parameters of the indicated preparations in male Wistar rats(n=
3.6.Residual solvent
ThesolventremaininginSDafterthepreparationprogresscan affect drug stability and is also potentially harmful to public health,depending on the solvent(s)employed[23,24].The residual ethanol was measured in the SM-SD prepared in the present study and the results were shown in Fig.7.The presence of a certain amount of ethanol(5000 ppm)is considered acceptablewithoutjustif i cationinpharmaceuticals.InSM-SDSE,residualethanolwasreducedfromalevelofapproximately 6000ppm(afterbeingdriedfor20minwithrotaryevaporation) toabout300ppmafterbeingdriedfor140min(Fig.7).However, the residue ethanol level was much lower in SM-SD-SEDS,at approximately1000ppmafter theinitial 20-mindryingperiod, and almost no residual ethanol after 140 min.These results indicated that more residual solvent was removed from SD generated using SEDS,rather than SE.
3.7.In vitro dissolution test
The dissolution prof i les shown in Fig.8 indicated that SM-SD exhibited faster drug release than the other preparations tested.As previously reported,rapid hydration of hydrophilic polymers promotes the drug wetting process in aqueous surroundings and enlarges the surface area of the solid-water interface,which may contribute to the improved SD dissolution rate[25].However,SM-SD-SEDS showed a cumulative release of more than 90%in the f i rst 5 min,whereas less than 70%release was observed from SM-SD-SE at 5 min.This may be due to the decreased particle size and reduced surface tension of the dissolution medium[26]when using supercritical f l uid technology.
3.8.Pharmacokinetics
Plasma SM was completely separated under the analytical conditions employed in this study.Silybin is an isomeric compound and two peaks were detected at about 18 and 21min.Thesumoftheareaofthetwopeakswasusedforthese pharmacokineticanalyses(Fig.9).Standard curveswerelinear (r=0.9996)overtherangeof0.08-3μg/ml.Theresultsobtained using this method indicated recoveries of 96.43±1.43%, 97.75±1.04%,and 97.93±1.46%at low,middle,and high concentrations,respectively.Thedetectionlimitwas30ng/ml.
Rat plasma SM was quantif i ed in vivo after oral administration of an aqueous suspension of SM or SM-SD-SEDS.These plasma prof i les were compared in Fig.10.The plasma SM concentration following administration of SM-SD-SEDS indicated more effective drug absorption,as compared with the SM aqueous suspension.All pharmacokinetic parameters (maximum plasma concentration[Cmax],time of Cmax[Tmax], area under the plasma concentration-time curve[AUC0-t], and plasma half-life[t1/2])were calculated individually for each subject in the group and the values were expressed as mean±standard deviation(n=6)(Table 2).
The AUC0-tof SM-SD-SEDS was 2.4-fold larger than that of SM suspension and this difference was statistically signif i cant (P<0.01).Cmaxwas also 1.9-fold higher and Tmaxoccurred earlier after oral administration of SM-SD-SEDS,as compared with SM aqueous suspension.These f i ndings indicated that SM-SD prepared by SEDS could provide a promising strategy for improving thebioavailabilityofSM.The enhanced bioavailability of SM delivered by SDs could be attributed to their improved rate of dissolution,which would facilitate drug absorption[27].
4.Conclusions
In summary,SM-SDs were successfully prepared using SEDS. The resultant SM-SD-SEDS considerably improved the drug dissolution prof i le and increased its oral absorption,as compared with SM powder.This study was innovative in its application of a new,high-eff i ciency,and environmentallyfriendly method to produce SDs and improve the dissolution and bioavailability for drugs with low aqueous solubility. Although future studies will be required to increase the oral bioavailability of SM further,the present study proved that supercritical f l uid technology provided a viable alternative means to prepare drug-loaded SDs.
Acknowledgements
This work was supported f i nancially by the Subject Chief Scientist Program(10XD14303900)from Science and TechnologyCommissionofShanghaiMunicipality,andthe Specialized Research Fund for the Doctoral Program of Higher Education of China(20123107110005).
REFERENCES
[1]Luper S.A review of plants used in the treatment of liver diseases:part 1.Altern Med Rev 1998;3:410-421.
[2]Pradhan SC,Girish C.Hepatoprotective herbal drug, silymarin from experimental pharmacology to clinical medicine.Indian J Med Res 2006;124:491-504.
[3]Mohamed ES,Gamal ES,Tarek MI.Comparison of early treatment with low doses of nilotinib,imatinib and a clinically relevant dose of silymarin in thioacetamideinduced liver f i brosis.Eur J Pharmacol 2001;670:593-600.
[4]Freedman ND,Curto TM,Morishima C.Silymarin use and liver disease progression in the hepatitis C antiviral longterm treatment against cirrhosis trial.Aliment Pharm Ther 2010;33:127-137.
[5]Dixit N,Baboota S,Kohli K,et al.Silymarin:a review of pharmacological aspects and bioavailability enhancement approaches.Indian J Pharmacol 2007;39:171-179.
[6]Giacomelli S,Gallo D,Apollonio P,et al.Silybin and its bioavailable phospholipid complex(IdB1016)potentiate in-vitro and in-vivo activity of cisplatin.Life Sci 2003;70:1447-1459.
[7]Shangguan MZ,Yi Lu,Qi JP,et al.Binary lipids-based nanostructured lipid carriers for improved oral bioavailability of silymarin.J Biomater Appl 2014;28:887-896.
[8]Panapisal V,Charoensri S,Tantituvanont A.Formulation of microemulsion systems for dermal delivery of silymarin. PharmSciTech 2012;13:389-399.
[9]Ahmed S,Mohammed E,Mohammad A.Complement activation assay and in vivo evaluation of silymarin loaded liver targeting liposome.J Life Med 2014;2:15-24.
[10]Sonali D,Tejal S,Vaishali T,et al.Silymarin-solid dispersions:characterization and inf l uence of preparation methods on dissolution.Acta Pharm 2010;60:427-443.
[11]Bobe K,Bobe CR,Suresh S.Formulation and evalution of solid dispersion of atorvastatin with various carriers. Pharmacie Globale(IJCP)2011;1:1-6.
[12]Das SK,Roy S,Kalimuthu Y.Solid dispersions:an approach to enhance the bioavailability of poorly water-soluble drugs. Int J Pharmacol Pharm Technol 2014;1:2277-3436.
[13]Won DH,Kim MS,Lee S,et al.Improved physicochemical characteristics of felodipine solid dispersion particles by supercritical anti-solvent precipitation process.Int J Pharm 2005;301:199-208.
[14]Juppo AM,Boissier C,Khoo C.Evaluation of solid dispersion particles prepared with SEDS.Int J Pharm 2003;250:385-401.
[15]Antonio T,Eva M,Miguel A.Precipitation of tretinoin and acetaminophen with solution enhanced dispersion by supercritical f l uids(SEDS).Powder Technol 2012;217:177-188.
[16]Chen ZQ,Liu Y,Zhao JH,et al.Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable selfmicroemulsifying drug delivery system.Int J Nanomedicine 2012;7:1115-1125.
[17]Kim MS,Jin SJ,Kim JS.Preparation,characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent(SAS)process. Eur J Pharm Biopharm 2008;69:454-465.
[18]Juan J,Garcia R,Paloma M,et al.Changed crystallinity of mebendazole solid dispersion:improved anthelmintic activity.Int J Pharm 2011;403:23-28.
[19]Han HK,Lee BJ,Lee HK.Enhanced dissolution and bioavailability of biochanin A via the preparation of solid dispersion:in vitro and in vivo evaluation.Int J Pharm 2011;415:89-94.
[20]Guo YS,Shalaev E,Scott S.Physical stability of pharmaceutical formulations:solid-state characterization of amorphous dispersions.TRAC-Trend Anal Chem 2013;49:137-144.
[21]Patrick JM,Alfred CFR,David EN,et al.Effect of temperature and moisture on the miscibility of amorphous dispersions of felodipine and poly(vinyl pyrrolidone).J Pharm Sci 2009;99:169-185.
[22]Sarode AL,Sandhu H,Shah N,et al.Hot melt extrusion for amorphous solid dispersions:temperature and moisture activated drug-polymer interactions for enhanced stability. Mol Pharmaceut 2013;10:3665.
[23]Deconinck E,Canfyn M,Sacre PY,et al.Evaluation of the residual solvent content of counterfeit tablets and capsules. J Pharmaceut Biomed 2013;81:80-88.
[24]Ke K,Wei XF,Bao RY,et al.Contribution of residual solvent to the nucleation and reinforcement of poly(vinylidene fl uoride).Polym Test 2014;34:78-84.
[25]Park YJ,Xuan JJ,Oh DH,et al.Development of novel itraconazole-loaded solid dispersion without crystalline change with improved bioavailability.Arch Pharm Res 2010;33:1217-1225.
[26]Park J,Cho W,Cha KH.Solubilization of the poorly water soluble drug,telmisartan,using supercritical anti-solvent (SAS)process.Int J Pharm 2013;441:50-55.
[27]Li SM,Liu Y,Liu T,et al.Development and in-vivo assessment of the bioavailability of oridonin solid dispersions by the gas anti-solvent technique.Int J Pharm 2011;411:172-177.
Abbreviations:HPMC,hydroxypropyl methylcellulose;PVP,polyvinyl pyrrolidone;SD,solid dispersion;SE,solution evaporation;SEDS, solution-enhanced dispersion by supercritical fl uids;SM,silymarin.
*Corresponding author.School of Pharmacy,Shanghai University of Traditional Chinese Medicine,No.1200,Cailun Road,Shanghai 201203,China.Tel./fax:+86 21 51322198.
E-mail addresses:npfeng@hotmail.com,npfeng@shutcm.edu.cn(N.Feng).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2014.12.001
1818-0876/©2014 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
 Asian Journal of Pharmacentical Sciences2015年3期
Asian Journal of Pharmacentical Sciences2015年3期
- Asian Journal of Pharmacentical Sciences的其它文章
- GUIDE FOR AUTHORS
- UPLC-MS/MS for the determination of azilsartan in beagle dog plasma and its applicationin a pharmacokinetics study
- Design and comparative in-vitro and in-vivo evaluation of starch-acrylate graft copolymer based salbutamol sulphate sustained release tablets
- Characterization of recrystallized itraconazole prepared by cooling and anti-solvent crystallization
- Enhanced bioavailability of rebamipide nanocrystal tablets:Formulation and in vitro/in vivo evaluation
- Liposomes for systematic delivery of vancomycin hydrochloride to decrease nephrotoxicity: Characterization and evaluation