
Liposomes for systematic delivery of vancomycin hydrochloride to decrease nephrotoxicity: Characterization and evaluation
2015-05-15 13:09:12JunliLiuZhonglnWngFuingLiJinhuGoLongmeiWngGuihuHung
Junli Liu,Zhongln Wng,Fuing Li,Jinhu Go,Longmei Wng, Guihu Hung,*
aThe School of Pharmaceutical Science,Shandong University,Ji'nan,China
bResearch Institue of Orthopaedic Trauma of PLA,Kunming General Hospital of Chengdu Military Command, Kunming,China
Liposomes for systematic delivery of vancomycin hydrochloride to decrease nephrotoxicity: Characterization and evaluation
Junli Liua,Zhonglan Wanga,Fubing Lib,Jinhua Gaoa,Longmei Wanga, Guihua Huanga,*
aThe School of Pharmaceutical Science,Shandong University,Ji'nan,China
bResearch Institue of Orthopaedic Trauma of PLA,Kunming General Hospital of Chengdu Military Command, Kunming,China
ARTICLEINFO
Article history:
Received 1 September 2014
Received in revised form
3 December 2014
Accepted 5 December 2014
Available online 20 December 2014
Biodistribution
Nephrotoxicity
Pharmacokinetic
Systematic delivery
Vancomycin hydrochloride
liposome
Vancomycin hydrochloride(VANH),the f i rst glycopeptide antibiotic,is a water-soluble drug for the treatment of acute osteomyelitis.Liposomal formulations of VANH have already been manipulated and characterized,which was a mean of increasing their therapeutic index,reducing their toxicity and altering drug biodistribution.One of the challenges for preparing VANH-Lips is their low encapsulation eff i ciency(EE).In the present study,we aim to improve the liposomal formulation of VANH for higher EE,longer systemic circulation,reduced nephrotoxicity and enhanced antimicrobial activities.Vancomycin hydrochloride-loaded liposomes(VANH-Lips)were formulated by the method of modif i ed reverse phase evaporation.Based on the optimization of formulation with orthogonal experimental design,the average drug encapsulation eff i ciency and the mean particle size of VANH-Lips were found to be 40.78±2.56%and 188.4±2.77 nm.In vitro drug release of VANH-Lips possessed a sustained release characteristic and their release behavior was in accordance with the Weibull equation.After intravenous injection to mice, the mean residence time(MRT)of VANH-Lips group was signif i cantly prolonged in vivo and the AUC value was improved as well compared with the vancomycin hydrochloride solution(VANH-Sol)group.Furthermore,the biodistribution results in mice showed that VANH-Lips decreased the accumulation of VANH in kidney after intravenous injection.In conclusion,VANH-Lips may be a potential delivery system for VANH to decrease nephrotoxicity in the treatment of osteomyelitis.
©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/ licenses/by-nc-nd/3.0/).
1.Introduction
Osteomyelitis is an infection of the bone and bone marrow, which is attributed to bacteria contamination result from trauma,surgery,orthopedic f i xation device implantation.In consequence it brings huge challenges to orthopedic surgeons [1].Regulartreatmentincludesthoroughdebridement, continuous perfusion,and intravenous application of sensitive antibiotic for 4-6 weeks.Based on complete removal of the lesion,effective systemic application of antibiotics is the necessity of the treatment of chronic osteomyelitis.Antibiotics are administered to control inf l ammation and to prevent exacerbation in chronic infections[2].However,the eff i cacy of antibiotics is severely limited by its poor penetration.Longterm and high-dose strategy is,thus,required to improve the therapeutic outcome[3].Although there are plenty of antibiotics and the current surgical techniques are promising, patients having osteomyelitis are still suffered from high recurrence rate.This is due to drug resistance induced by frequent administrationsofsensitive antibiotics.Tobe treated clinically,therefore,osteomyelitis has been normally considered as one of the most obstinate orthopedic and postoperative complications[4].
As the f i rst glycopeptide antibiotic,VANH was developed in the 1950s[5].Evenif the concentration ofvancomycinis low,it is still valid against the majority of Gram-positive bacteria,for example,Staphylococcus aureus and other staphylococcus species[5].S.aureus is one of the most frequent pathogenic bacterium of osteomyelitis[6].Although it works specif i cally on bacterial cell wall peptidoglycan,but,the side effects, includingmostnotablynephrotoxicity,ototoxicity,and neuromuscular blockade,appeared in the early course of its application.These side effects limit its wide application[5,7].
Renaleliminationofvancomycinismainlythrough glomerular filtration and partly mediated by active tubular secretion[8].Most of VANH is removed unchanged in the urine[9].Renal dysfunction is a major omen of vancomycin treatment failure[10].Gene expression prof i ling studies in animals with high-dose VANH suggest that cellular necrosis resulted from VANH accumulation in proximal tubular cells may be the underlying mechanism of nephrotoxicity[11]. Many of previous reports has demonstrated a highly signif icant relationship between vancomycin dose and the time to nephrotoxicity[12,13].In-patients who suffer from chronic osteomyelitis often have low immunity.Therefore it easily leads to chronic kidney disease and acute kidney injury.In addition,Clinical failure reasons have also been reported owing to its slow,time-dependent bactericidal activity,poor intracellular penetration into macrophages[14].Therefore, merely increasing the parenteral treatment doses of vancomycin offers an essentially increased risk of drug toxicity that is not offset by any appreciable benef i t[15].
Liposomes have been proven to be a hopeful drug delivery system for a variety of drugs including antibiotics[16].As their lipid bilayer are made up of phospholipid and cholesterol, which is similar to the cell membrane and can readily fuse with infectious microbes,Liposomes are considered to be a non-toxic and biodegradable drug carrier for sustained and targeted delivery.They are also the most widely used antimicrobial drug delivery vehicles[17].Encapsulation of antibiotics in liposomes is known to enhance their antimicrobial activities while minimizing their toxic effects[7].In addition,the sustained release of antibiotics from the liposomes may prolong the half-lives of these drugs in the body.
Encapsulation of VANH into liposomes has been attempted by our group as well as several other investigators as a mean of increasing their therapeutic index,reducing their toxicity and altering drug biodistribution[14,15,18-20].Nevertheless, due to the highly hydrophilic property,one of the challenges preparing VANH-Lips is their low encapsulation eff i ciency.If so,this will require high amount of liposomal formulations to achieve satisfactory therapeutic dose,a reason that prone to drug resistance.In the present work,several methods have been attempted for this challenging endeavor.The EE of the liposomes prepared by Andrew Pumerantz[15]and Krishna Muppidi[14]are both 9%.The EE for vancomycin hydrochloride into liposomes preparedby thin f i lm dispersion methodis 2.34%[19].Yang DM[20]prepared the multivesicular liposomes,whose mean EE and particle size is 24.9%and 3.3 μm, respectively.The EE of cationic liposomes prepared by Tao Ma [18]is 7.58%.The objective of this study was to prepare vancomycin-encapsulated liposomes with relatively high encapsulation eff i ciency and performed an in vivo study using healthy mice to compare their pharmacokinetic and biodistribution prof i les with those of mice treated with the standard vancomycin solution.The optimal liposomes prepared by modif i ed reverse phase evaporation methods were composed of injectable soybean phosphatides,cholesterol and VANH.The composition of the formulation is non-toxic, and the preparation process is simple and controllable.The optimal formulations with appropriate drug encapsulation percentage and homogenous particle size distribution were selectedtoinvestigatethepharmacokineticsandbiodistribution after intravenous administration,utilizing mice as animal models compared with free vancomycin hydrochloride,in order to provide the basic knowledge for further development and evaluation of liposome-encapsulated VANH formulation.
2.Materials and methods
2.1.Materials
Soybean lecithin(Injection grade,phosphatidylcholine accounts for 95%pH 5.0-7.0)was provided by Shanghai Taiwei Pharmaceutical Co.,Ltd,Shanghai,China.Cholesterol was purchased from Shanghai Medical Chemical Reagent Co.,Ltd. (China).VANH was purchased fromShanghai ziyi ReagentCo., Ltd.(China).The other chemicals were of analytical reagent grade or higher.
Animal:Kunming strain mice(weighed between 18 and 22 g,femaleor malewere providedby the MedicalAnimalTest Center of Shandong University)were used for the in vivo pharmacokinetics and biodistribution studies.The animals were fasted 12 h before drug administration.The animal experiment protocol was reviewed and approved by the Institutional Animal Care and Use Committee of Shandong University.
2.2.Preparation of VANH-Lips
Formulation of VANH-liposomes were prepared using various methods.
2.2.1.Freeze-thaw method[21]
Pre-formed empty liposomes were prepared by the f i lmhydration method.Brief l y,lipid(soybean lecithin and cholesterol)was mixed with chloroform and evaporated using a rotary evaporator(Shanghai Yarong Instrument Co.,Shanghai, China)at an elevated temperature(40°C).During evaporation, alowvacuumwasappliedinitiallytoavoidburstingofthelipid solution.The dried lipid was placed under vacuum overnight to remove residual solvent.Then VANH-Sol was added to hydrate for 10 min and the mixture was frozen at-20°C for 20 min.The liposomes were then sonicated at 5 min followed by two freeze-thaw cycles(-20°C for 20 min,45°C for 10 min).
2.2.2.Proliposome method
Def i ned amounts of phospholipids and cholesterol were weighed into a round-bottom f l ask and dissolved in chloroform,forming a transparent solution,which was described by Isailovic et al.[22].Prescribed amount of sorbitol powder was added in the solution.The organic solvent was then removed under reduced pressure by the rotary evaporator to form proliposome.Afterwards,VANH-Sol was poured into the proliposome solution,and the mixture was sonicated to form uniform liposome.
2.2.3.Remote loading method by ammonium sulfate gradients
This method included two steps[23].The f i rst step was the formation of empty liposomes.The liposomes were prepared by dissolving phospholipids/cholesterol in chloroform.The organic solvent was then removed in a 40°C water bath under reduced pressure by the rotary evaporator to form lipid membrane.The lipid membrane was then hydrated in ammonium sulfate solution.And the mixture was sonicated to form translucent solution.Taken suitable amounts of liposomes in dialysis bag,and dialysis in distilled water for 12 h at room temperature,and the empty liposomes were prepared.The second step was drug loading.Brief l y,take empty liposomes,adding to VANH-Sol,and incubate in water bath,then the drugs could across the lipid bilayer and were entrapped into the vesicles.
2.2.4.pH-gradient method
Suitable amounts of phospholipids and cholesterol were weighed into a round-bottom f l ask and dissolved in chloroform,as described by Zhou[24].Then chloroform was removed in a 40°C water bath under reduced pressure by the rotary evaporator to form lipid membrane.The lipid membranewasthenhydratedinthecitricacidsolution(300 mmol/L)to produce multivesicular vesicles.The liposomes were then homogenized at 800-1000 bar.In order to form the transmembrane pH-gradient between the external phase and internal phase of liposome,Na2HPO4solution (0.5 mol/L)was added into the system to adjust the external pH of liposomes to 6.5.Then,the system was incubated under 50°C for 10 min.
2.2.5.Reverse phase evaporation method
Appropriate amounts of phospholipids and cholesterol were weighed into a round-bottom f l ask and dissolved in chloroform[25].Subsequently,VANH-Sol was directly added to the dispersed solution of phospholipids,and the mixture was sonicated 3 min at room temperature in an ultrasonic bath (Kun Shan Ultrasonic Instruments Co.,Ltd).The organic solvent was then removed in a 40°C water bath under reduced pressure by the rotary evaporator.Suff i cientamounts of water was added to hydrate for an additional 10 min.Then,the liposomal emulsion was maintained under control temperature to make the liposome structure more stable.
2.2.6.Modif i ed reverse phase evaporation-rehydration method
Similar to reverse phase evaporation method,except for the difference that VANH-Sol was dispersed with a microinjector (Kd Scientif i c 781100,Fabrique'Auxetats-Units Co.Ltd.,USA) into the dispersed solution of phospholipids.And the mixture was sonicated to form a primary emulsion at room temperature in an ultrasonic bath.
2.3.Optimization of formulation with orthogonal experimental design
On the basis of preliminary test,modif i ed reverse phase evaporation method reached the highest EE.Four selective formulation factors that mainly affected EE were chosen as research objects,including(A)the ratio of cholesterol to lecithin(w/w),(B)the ratio of drug to lipids(w/w),(C)the ratio of water phase to oil phase(v/v)and(D)hydration temperature. In this place,water phase is VANH-Sol and oil phase is the chloroform solution of phospholipid and cholesterol.Nine formulations were designed for test according to L9(34) orthogonal experimental design,in order to screen the optimal formulation.On the basis of size plus EE as the evaluation index,the factors and levels of the orthogonal experimental design are listed in Table 1.
2.4.Freeze-drying of VANH-Lips
In the freeze-drying process,mannitol(5%,w/v)was used as a cryoprotectant.First the fresh prepared VANH-Lips with 5% mannitol were pre-frozen using an ultra-cold freezer(MDF-382E,Sanyo Electric Co.,Ltd.,Osaka,Japan)for 24 h at-80°C. Then,theresultantsamplesweretransferredtothelyophilizer(FD5-2.5,GOLD SIM,Issaquah,WA)at-50°C for 48 h.The lyophilized powder was collected for further experiments.

Table 1-Factors and levels of the orthogonal experimental design.
2.5.Characterization of VANH-Lips
2.5.1.Visualization of liposomes by transmission electron microscopy(TEM)
TEM(H-7000,Hitachi,Japan)was used as a visualizing aid for vesicles.Samples were negatively stained with a 2%aqueous solution of phosphotungstic acid.Vesicular suspension samples were dried on a carbon-coated grid for staining.The excess solution was removed by blotting.After drying,the specimen was viewed under the TEM.
2.5.2.Measurement of particle size and pH value
Measurements were performed using a Beckman Delsa™Nano C Particle Analyzer(Beckman Coulter A53878,Otsuka Blectronics Co.Ltd.,USA).Samples were placed in plastic disposable cuvettes and equilibrated at 25°C.Particle size measurements included z-average,PDI,and PDI width.The pH value of VANH-Lips was determined with a digital pH meter(FE20,Mettler Toledo,Greifensee,Switzerland).Each measurement was made at least in triplicate at 25°C.
2.5.3.Determination of encapsulation eff i ciency(EE)
The EE of VANH-Lips were determined by ultraf i ltration-HPLC method using an Agilent G1310A pump and an Agilent G1314A variable wavelength detector set at 230 nm.VANH was monitored at a wavelength of 230 nm with the column, InertSustain®-C18 column(4.6 mm×250 mm)(Shimazu-GL, JAPAN).The mobile phase was composed of 0.05 mol/L potassium phosphate monobasic monopotassium phosphate solution(pH 3.2)and methanol(spectroscopic grade)(78:22, ml/ml)at a f l ow rate of 1.0 ml/min.As follows,0.1 ml of liposome was mixed with 2 ml methanol,adding distilled water volume to 10 ml.The total drug contents in liposomes were measured by HPLC.0.1 ml of liposome was mixed with 0.5 ml distilled water into ultraf i ltration centrifuge tube (MILLIPORE,UFC901024 15 M 10 K),and was centrifuged at 4000 rpm for 40 min(3K30,Sigma,Germany).Then another 200 μl distilled water was added into the mixture and centrifuged at 4000 rpm for 20 min again.The free drug content in the outer tube was also measured by HPLC.The EE and DL were calculated by using under-mentioned equations(1)and (2)respectively.
wherewtotalwas the analyzed weightof drugin liposomes,wfreewas the analyzed weight of free drug,wlipidwas the weight of lipid added in system.
2.5.4.The drug release experiment in vitro from VANH-Lips The in vitro release study of VANH-Lips was carried out at 37°C using dynamic dialysis technique[26,27].The liposomes with encapsulation eff i ciency 40.31%were used for this analysis.Brief l y,the VANH-Lip dispersion(2 ml,donor solution)waskeptinadialysismembrane(MD34, 8000-14,000,AMERICA)with a molecular weight cut-off of 8000-14,000 Da and this system was immersed in 30 ml of pH 7.4 buffer solution.The medium was kept at 37°C under continuous magnetic stirring of 100 rpm.At a regular interval of time,0.5 ml of receiver solution was withdrawn and same volume of fresh medium was replaced.The VANH-Sol obtainedfrom a dialysis testwas usedas control.The percentage of drug released was determined using the HPLC conditions mentioned previously.The mean calculated values were obtained from 3 replicates.The cumulative fraction of release rate was calculated from the following equation and the results are expressed as mean±standard deviation(SD):
cnis the drug concentration in the release medium of each time interval,v0is the total volume of the release medium,viis the volume of the withdrawn medium,ciis the drug concentration in the release medium at time.
2.6.Pharmacokinetics studies and drug distribution studies in mice
Kunming strain mice,weighing between 18 and 22 g,were randomly divided into two groups.Group 1 was treated with VANH-Sol while group 2 was treated with the VANH-Lips.The concentration of VANH in liposome is 0.96 mg/ml,while the VANH was dissolved in the sterile water for injection to obtain the same concentration.Each preparation was injected through the tail vein at the VANH dose of 15 mg/kg mouse. Blood samples were taken from the terminal retro-orbital bleeding at various times(0.083,0.25,0.5,1,2,3,4,6,8 and 10 h)into micro-tubes containing sodium heparin as an anticoagulant,andcentrifugedimmediately(10min, 12,000 rpm).0.2 ml 10%zinc sulfate solution was added to an aliquot(0.2 ml)of each plasma sample and mixed for 3 min by vortex to extract VANH.Following centrifugation at 12000 rpm for 10 min,the organic phase was transferred to a glass tube and the solvent was evaporated under nitrogen stream at 40°C.The dry sample was then dissolved in 100 μl mobile phase and 20 μl of the solution was injected into the HPLC column to measure VANH peak area and calculate its concentration by standard curve method.
The heart,liver,spleen,lungs,kidneys and brain of each mouse was rapidly excised following blood collections,and immediately washed twice with normal saline(0.9%NaCl), wipedwith f i lter paper,weighedand homogenized with 1.0 ml normal saline(0.9%NaCl),except for liver(2 ml).0.2 ml 10% zinc sulfate solution was added to an aliquot(0.2 ml)of each plasma sample and mixed for 3 min by vortex to extract vancomycin hydrochloride.After vortexing(IKA®VORTEX1, GEMANY)for 3 min,the sample was centrifuged for approximately 10 min at 12,000 rpm.The supernatant was collected and the solvent was evaporated under nitrogen gas at 40°C. The dry sample was reconstituted in 100 μL mobile phase for measurement of VANH by HPLC.
The concentration of VANH was measured by the method of reversed-phase HPLC with the column,InertSustain®-C18column(4.6 mm×250 mm).VANH was monitored at a wavelength of 230 nm.The mobile phase was composed of 0.05 mol/L potassium phosphate monobasic monopotassium phosphate solution(pH 3.2)and methanol(spectroscopic grade)(82:18,ml/ml)at a f l ow rate of 1.0 ml/min.Aliquots of deposed supernatants(20 μL)were loaded on the HPLC.The detection limit of VANH was 0.4 ng.Intraday and interday variabilities were less than 3%and 5%,respectively.Mean recovery rates of each organ exceeded 80%,RSD<5%.
2.7.Statistical analysis
Data were expressed as mean±standard deviation(SD).The statistical analysis of signif i cance among various treatments was performed using unpaired Student's t-test with P<0.05 indicating signif i cant difference.An analysis of variance (ANOVA)test was also used if necessary.Pharmacokinetic parameters of VANH-Lip were obtained using DAS2.0(drug and statistics for windows)program.
3.Results and discussions
3.1.Encapsulation eff i ciency(EE)of VANH-Lips
VANH is highly hydrophilic and water soluble.According to the results showed in previous report[14,15,18-20],the EE of liposome-loaded water-soluble drugs is low.There are many approaches for the preparation liposomal formulation of VANH including thin f i lm dispersion method[19],double emulsion method[20]and reverse phase method[18].So it is very difficult to prepare high EE,stable VANH-Lips by classic passive loading method due to the fact that VANH is hydrosoluble.
So we attempt different ways to improve the EE,including freeze-thaw method,proliposome method,modif i ed reverse phaseevaporation-rehydrationmethod,remoteloading methods by ammonium sulfate gradients and pH-gradient method.Modif i ed reverse phase evaporation method,is used to prepare liposomes with a large internal aqueous space,which is advantageous to encapsulate water-soluble drugs [25].Based on reverse phase evaporation method described by Szoka and Papahadjopoulos[25],the difference was that VANH-Sol was uniformly dispersed with a microinjector to make the liposome with better formability,emulsif i cation time was increased to reduce the size of VANH-Lips,and the hydration time was shorten to improve the EE.
Modif i ed reverse phase evaporation method can make the drug dispersed more uniform.Then a more uniform f i lm was formed after rotary evaporation and the particle size was smaller after hydration.Thus we can reduce ultrasonic time after hydration to avoid leakage of drugs,making drug encapsulation eff i ciency improved.
According to the results showed in Fig.1,the EE of the liposome preparedby freeze-thawn method is the highest,the second is modif i ed reverse phase evaporation method.In the preparation of liposomes,freeze-thaw cycling is implemented to reduce the lamellarity of liposomes,form a less polydispersedsystemand/ordisrupt the liposomal bilayer to allow drug molecules to diffuse into the liposome,promoting encapsulation.Three batches of liposomes were prepared by freeze-thaw method.The particle size of the three batches were 321.6,457.7,489.5 nm,respectively.The mean size was 422.9 nm,and RSD is 21.09%.So the size of the liposome prepared by freeze-thaw method is diff i cult to control,and the reproducibility is poor.Regarding the eff i ciency of drug encapsulated as shown in Fig.1,correlating to the different preparation methods,the mean encapsulation percentage of VANH prepared by modif i ed reverse phase techniques was elevated signif i cantly and the particle size distribution was more homogeneous as compared to that of thin f i lm or proliposome method.However,it should be noted the f i ne drug encapsulation eff i ciency of liposome was obtained using 1:15 M ratio of VANH and phospholipids.Therefore,modif i ed reverse phase evaporation method is the best choice.
3.2.Results of the test of orthogonal design of VANH-Lip
Based on the preliminary experimental results,the EE can be improved through formulation and preparation optimization. On the basis of studying many documents,four selective formulation factors that mainly affected EE and size were chosen as research objects,including(A),(B),(C)and(D). Furthermore,the formulation factors affecting EE were studied in orthogonal experimental design.The resultsare showed in Table 2.
The results were calculated by range analysis.According to Table 2,the effect of(A)the ratio of cholesterol to lecithin (w/w)was extremely signif i cant;the effect of other factors isnot signif i cant.The range ref l ected the extent of each factor affecting on index and range was bigger,extent affected was greater.The effects of each factor on the EE were as follows: (A)the ratio of cholesterol to lecithin(w/w)>(B)the ratio of drug to lipids(w/w)>(D)hydration temperature>(C)the ratio of water phase to oil phase(v/v).K1,K2,K3 represented the sum value of each level.On the basis of the synthetic mean value of a certain factor(A or B or C or D),the optimized level combination of each factor was chosen.The lower the synthetic mean value,the better the level was and a higher EE can be achieved in this level.Analytical results of three factors were A:1>2>3;B:1>2>3;C:1>2>3;D:1>3>2,so the optimal parameters is A3B3C3D2.Furthermore,the encapsulation percentage was elevated accompanied with the increment of phospholipid amount.So the optimal formulation is adjusted slightly to improve drug loading eff i ciency,precisely the ratio of cholesterol to lecithin is 1:6.5,the ratio of drug to lipids(w/w)is 1:15,hydration temperature 45°C,the ratio of water phase to oil phase(v/v)is 1:4.The EE of the liposome prepared by the optimum formulation reached to 40.78%.The optimum formulation is feasible and the EE of the liposome prepared by the optimum formulation is higher.Furthermore, with the optimization of formulation,the achievement of liposomes with higher EE by reverse phase evaporation method would be helpful to improve pharmaceutical and pharmacological effects in curing Osteomyelitis.In short,the reverse phase evaporation method was superior to previous reports in encapsulating vancomycin hydrochloride.
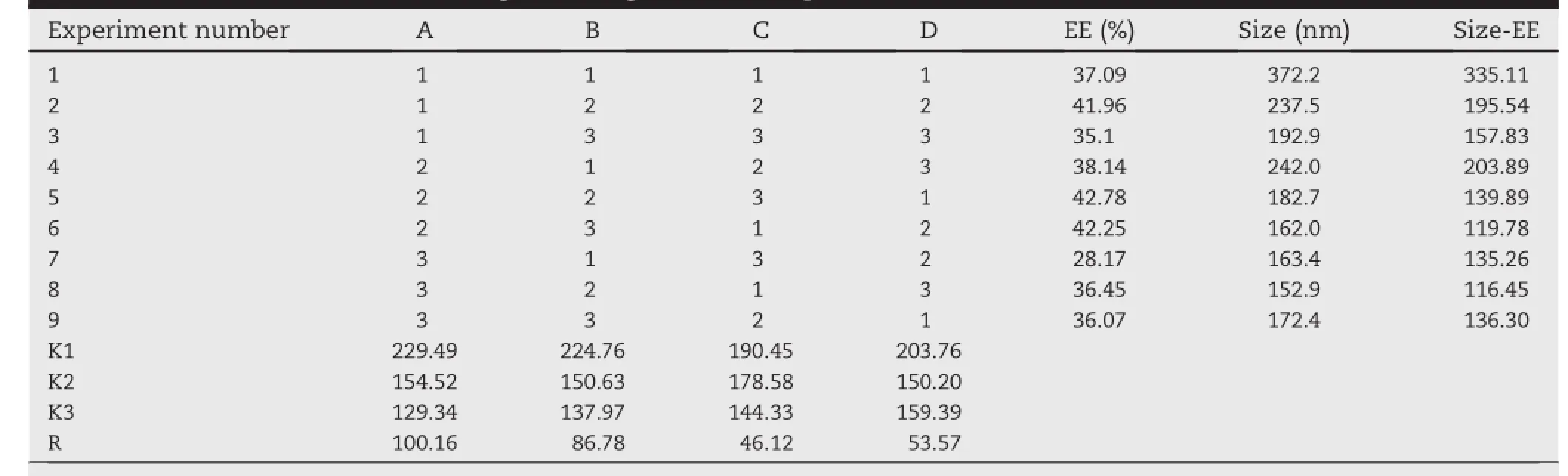
Table 2-Results of the test of orthogonal design of VANH-Lips.
3.3.Characterization of VANH-loaded liposome
3.3.1.Morphology and physicochemical property of VANHLips
The photographs of fresh-prepared VANH-Lips,the VANHLips freeze-dried powder and the suspension of VANH-Lips freeze-dried powder(dispersed with distilled water)are shown in Fig.2.The particle size of the fresh-prepared VANHLips was 188.4 nm,indicating light blue opalescence.The lyophilized powder of VANH-Lips appeared as full,no collapse of the porous solid block.The lyophilized powder displayed good redispersibility and the reconstituted VANH-Lips with distilled water were still an opalescent colloidal solution.The morphology of VANH-Lips was observed by TEM and the observation results were displayed in Fig.3.The TEM image shows that most liposomes were spherical particles with approximate size and uniform dispersion.
As shown in Table 3,the EE and drug loading of the freshprepared VANH-Lips Table 3 were 40.78%and 2.55%,which wererelativelyhigherthanthatreportedpreviously [14,15,18-20].The pH of VANH-Lips was 5.96,which is consistent with the requirements for intravenous injection.After lyophilization,the EE and drug loading were slightly reduced to 35.58%and 2.23%.This is possibly because that VANH is so hydrophilic that it is easy to leak from the liposome in the freeze-drying process.

Table 3-Physicochemical characteristics of VANH-Lips:(A)fresh-prepared,(B)the suspension of lyophilizedpowder,each data was expressed as mean±SD(n=3).
3.3.2.In vitro drug release of VANH-Lips
In order to mimic the behavior of liposomes after intravenously administration to patients,VANH-Lips were incubated usinga dialysismembranein pH7.4 phosphatebuffersolution at 37°C.The release experiment was carried out under sink conditions.Theaccumulativereleasepercentagepro fi le versus time of VANH-Lips and VANH-Sol is shown in Fig.4. The release pro fi le of VANH from VANH-Lips was,further, compared with VANH-Sol.It was apparent that almost 75%of the drug released within 4 h of dialysis,when VANH-Sol was dialyzed in pH 7.4 phosphate buffer solution.The VANH release from VANH-Lips,however,did not show signi fi cant burst release.VANH was released slower from VANH-Lips than VANH-Sol obviously.The VANH release from VANHLips was relatively slow and controlled.The release of VANH from VANH-loaded liposomes was in accordance with the Weibull equation and can be expressed using the following equation:ln[1/(1-R/100)]=0.4742lnt-0.9485,r=0.9791.The release of VANH from VANH-Sol was in accordance with the fi rst-order kinetic model and can be expressed using the following equation:ln(100-R)=-0.2722t+4.4234,r=0.9951. This equation can be applied to forecast the release amount of VANH at different time,or calculate the time to release a certain amount of VANH,which could predict the status of liposomes in practical applications.The release prof i le of VANH from liposomes was biphasic.The initial fast release of around 31%of the drug from the liposomes was observed in the f i rst 1 h,which could probably be due to the portion of the drug that leaked out of liposomes and the unloaded drug.
3.4.Pharmacokinetics studies and drug distribution studies in mice
3.4.1.The experimential results of pharmacokinetics
At the range of 0.5-40 μg/ml,the standard curve was A=14.699C+0.1388 and the standard showed a good linearity with a correlation coeff i cient of 0.9999.The blood concentration-time curves of VANH in mice after intravenous administration of a single 15 mg/kg dose of VANH-Sol and VANH-Lips were shown in Fig.5.The pharmacokinetic parameterscalculatedbyDAS2.0softwarewereshowninTable4. Based on the analysis of the models and parameters,it was concluded that the mean blood concentration-time prof i les for both VANH-Sol and VANH-Lips were corresponded to thetwo-compartment model following intravenous administration,the weight coeff i cient was expressed as 1/cc.

Table 4-Main pharmacokinetic parameters of VANH in mice after intravenous administration of VANH-Sol and VANH-Lips at a single dose of 15 mg/kg.
After i.v.administration,VANH-Sol was removed from the blood circulation of mice with a half-life of about 1.888 h.On the contrary,the half-life of VANH-Lips increased to 2.240 h comparedwithVANH-Sol.Furthermore,comparedwith VANH-Sol,MRT0-tand MRT0-∞of VANH-Lip increased by 0.83 and 0.61 fold,respectively.The extended MRT may due to the sustained release of VANH from VANH-Lips and the fact that the reticuloendothelial system(RES)removal could be avoided [28].What'smore,AUC0-tandAUC0-∞ofVANH-Lips increased about 1.13-fold and 0.99-times compared with those of VANH-Sol,respectively.Therefore,the prolonged blood circulation and higher AUC value of VANH-Lips may contribute to higher therapeutic eff i cacy on long-periodic treatment of osteomyelitis.
The plasma concentration-time prof i le after intravenous administration of VANH-Sol and VANH-Lips at a dose of 15 mg/kg VANH was shown in Fig.5.The measured VANH plasma concentration achieved from VANH-Lips group was higher than that of the control VANH-Sol at each time point. ThentheplasmaconcentrationofVANH-Soldeclined signif i cantly more rapid than that of VANH-Lips.This is possibly because that VANH was encapsulated into phospholipid bilayer and released into the plasma for a prolonged period of time.Moreover,it was reported that nanoparticles smaller than 220 nm could escape macrophage clearance to some extent.So the VANH-Lips can reduce clearance by macrophage as well,which will contribute to long circulation in blood[28].Pharmacokinetic results in this study indicated VANH encapsulated in liposomes retains in the blood stream much longer than the VANH-Sol.So the VANH-Lip has more time to interact with the APL cell,which may benef i t to the long-period clinical application of VANH.

Table 5-Biodistribution of vancomycin in liver,spleen and kidney tissue after intravenous administration of a dose (15 mg/kg)of solution and liposome formulations.
3.4.2.Drug distribution studies
Within 0.2-30 μg/ml VANH concentration,the standard curves of all measured organs were established and the correlation coeff i cients range from 0.9990 to 0.9997.The optimum liposome-loaded VANH formulation by modif i ed reverse phase evaporation method was used to investigate the biodistribution of liposomal VANH in mice and evaluate its relative retaining activities to the certain organs such as heart, liver,spleen,lung,kidney and brain by determining the concentration of vancomycin hydrochloride.The tissue distributions of VANH after intravenous injection of a single 15 mg/kg dose of VANH-Sol and VANH-Lips were shown in Fig.6, respectively.Remarkably,the drug distribution was different in the VANH-Lips group and the VANH-Sol group.Brief l y,the concentration of VANH in kidney,an important excretory organfor i.v.deliveryof VANH,was muchlowerfor theVANHSol group,which may be expected to reduce or avoid the potential side effects to kidney.Furthermore,VANH-Lips resulted in a higher drug accumulation in liver and spleen compared with VANH-Sol(P<0.05),which may be explained by the fact that lipsome presented accumulative activity in RES sites such as lung,liver and spleen[29].Conversely the biodistribution of VANH-Lips in non-RES sites such as in kidney decreased compared with VANH-Sol,which potentially resulted in the reduction of renal damages.
Notably,as is shown in Fig.7,the drug concentrations in kidey were decreased tremendously after the administration of VANH-Lips compared with the VANH-Sol,which could improve the eff i ciency of VANH and decrease the side effects. This phenomenon may be explained by the fact that lipsome encapsulation results in a decrease of the renal clearance[30].
The cmax,AUC and MRT values for the two groups in different tissue were calculated and are reported in Table 5. The tissue distribution results were evaluated according to Re(Re=AUCVANH-Lip/AUCVANH-Sol).If the value of Re exceeds 1, the tissue is exposed to drug to a greater extent by theliposomes[31].The Re of heart,liver,spleen,lung and kidney were 1.040,2.167,1.982,1.155,0.645 and 0.750,respectively, shown in Table 5.It is in accordance with Fig.6.It demonstrated that liposomes presented accumulative activity in reticuloendothelial system(RES)sites such as spleenand liver. In addition,the value of Re for lung were also higher than 1, which showed that VANH-Lips were also inclined to distribute to lung though less preferable to liver and spleen.Therefore VANH-Lips performed their potential as a delivery system targeting to lung for the treatment of pulmonary inf l ammation.The AUC of heart and brain were not signif i cantly changed.Conversely,the biodistribution of liposomes in non-RES sites such as in kidney decreased with descending Re compared with VANH-Sol,which potentially resulted in the reduction of nephrotoxicity or renal damages.This prompted the prepared VANH-Lips had the advantage in the longperiodic treatment of osteomyelitis.
4.Conclusions and outlook
In thiswork,VANH-Lipswassuccessfully developed.The EE of VANH-Lips were signif i cantly increased.The in vitro and in vivo physicochemical properties of VANH-Lips were characterized and evaluated in detail.The in vitro drug release experiments exhibited biphasic drug release characteristic and slower release than VANH-Sol.The pharmacokinetics studies conf i rmed that the circulation time of VANH in blood can be extended by liposome encapsulation.The tissue distribution study indicated that VANH-Lips decreased the drug accumulation in kidney following intravenous injection in mice.We,therefore,conclude that VANH-Lips may serve as a promising carrier for systematic delivery of VANH in the treatment of osteomyelitis.
Liposome as drug carriers signif i cantly inf l uence on drug distribution and reduce toxic side effects during antibiotic therapy[32-34].In this study,VANH-Lips were found to reduce the distribution of VANH in kidney,which might help to lower its renal toxicity and eliminate its potential toxicity of kidney tubules.These f i ndings suggested that VANH-Lips might have a potential to be developed as a promising pharmaceuticalpreparationforthetreatmentofchronic osteomyelitis.
Acknowledgements
The authors would like to express thanks to the School of Pharmaceutical Science,Shandong University for providing the required infrastructure to carry out the study.The authors thank Miao Lei for her assistance.
REFERENCES
[1]Wu P,Grainger DW.Drug/device combinations for local drug therapies and infection prophylaxis.Biomaterials 2006;27:2450-2467.
[2]Stengel D,Bauwens K,Sehouli J,et al.Systematic review and meta-analysis of antibiotic therapy for bone and joint infections.Lancet Infect Dis 2001;1:175-188.
[3]Hatzenbuehler J,Pulling TJ.Diagnosis and management of osteomyelitis.Am Fam Physician 2011;84:1027.
[4]Liu XM,Zhang Y,Chen F,et al.Prevention of orthopedic device-associated osteomyelitis using oxacillin-containing biomineral-binding liposomes.Pharm Res 2012;29:3169-3179.
[5]Reynolds PE.Structure,biochemistry and mechanism of action of glycopeptide antibiotics.Eur J Clin Microbiol Infect Dis 1989;8:943-950.
[6]Yang CJ,Li Q,Wu GC,et al.A practical model of osteomyelitis-induced bone pain by intra-tibial injection of Staphylococcus aureus in rats.Neurosci Lett 2012;513:198-203.
[7]Alipour M,Halwani M,Omri A,et al.Antimicrobial effectiveness of liposomal polymyxin B against resistant Gram-negative bacterial strains.Int J Pharm 2008;355:293-298.
[8]Nakamura T,Takano M,Yasuhara M,et al.In-vivo clearance study of vancomycin in rats.J Pharm Pharmacol 1996;48:1197-1200.
[9]Rybak MJ.The pharmacokinetic and pharmacodynamic properties of vancomycin.Clin Infect Dis 2006;42:S35-9.
[10]Hidayat LK,Hsu DI,Quist R,et al.High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections:eff i cacy and toxicity.Arch Intern Med 2006;166:2138-2144.
[11]Dieterich C,Puey A,Lyn S,et al.Gene expression analysis reveals new possible mechanisms of vancomycin-induced nephrotoxicity and identif i es gene markers candidates. Toxicol Sci 2009;107:258-269.
[12]Lodise TP,Lomaestro B,Graves J,et al.Larger vancomycin doses(at least four grams per day)are associated with an increased incidence of nephrotoxicity.Antimicrob Agents Chemother 2008;52:1330-1336.
[13]Lodise TP,Patel N,Lomaestro BM,et al.Relationship between initial vancomycin concentration-time prof i le and nephrotoxicity among hospitalized patients.Clin Infect Dis 2009;49:507-514.
[14]Betageri G.Development and stability studies of novel liposomal vancomycin formulations.ISRN Pharm 2012;2012.
[15]Pumerantz A,Muppidi K,Agnihotri S,et al.Preparation of liposomal vancomycin and intracellular killing of meticillinresistant Staphylococcus aureus(MRSA).Int J Antimicrob Agents 2011;37:140-144.
[16]Drulis-Kawa Z,Dorotkiewicz-Jach A.Liposomes as delivery systems for antibiotics.Int J Pharm 2010;387:187-198.
[17]Huh AJ,Kwon YJ.“Nanoantibiotics”:a new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era.J Control Release 2011;156:128-145.
[18]Ma T,Shang B-C,Tang H,et al.Nano-hydroxyapatite/ chitosan/konjac glucomannan scaffolds loaded with cationic liposomal vancomycin:preparation,in vitro release and activity against Staphylococcus aureus biof i lms.J Biomater Sci Polym Ed 2011;22:1669-1681.
[19]Kadry AA,Al-Suwayeh SA,Abd-Allah AR,et al.Treatment of experimental osteomyelitis by liposomal antibiotics. J Antimicrob Chemother 2004;54:1103-1108.
[20]Yang DM,Xu YQ,Li FB,et al.Preparation of cationic vancomycin hydrochloride multivesicular liposomes and its quality.Chin J Reparative Reconstr Surg 2013;27:444.
[21]Costa AP,Xu X,Burgess DJ.Freeze-anneal-thaw cycling of unilamellar liposomes:effect on encapsulation eff i ciency. Pharm Res 2014;31:97-103.
[22]Isailoviˊc BD,Kostiˊc IT,Zvonar A,et al.Resveratrol loaded liposomes produced by different techniques.Innov Food Sci Emerg Technol 2013;19:181-189.
[23]Zucker D,Marcus D,Barenholz Y,et al.Liposome drugs' loading eff i ciency:a working model based on loading conditions and drug's physicochemical properties.J Control Release 2009;139:73-80.
[24]Zhou Y,Wei Y,Liu H,et al.Preparation and in vitro evaluation of ethosomal total alkaloids of Sophora alopecuroides loaded by a transmembrane pH-gradient method.AAPS PharmSciTech 2010;11:1350-1358.
[25]Szoka F,Papahadjopoulos D.Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation.Proc Natl Acad Sci 1978;75:4194-4198.
[26]Santos SS,Lorenzoni A,Ferreira LM,et al.Clotrimazoleloaded Eudragit®RS100 nanocapsules:preparation, characterization and in vitro evaluation of antifungal activity against Candida species.Mater Sci Eng C 2013;33:1389-1394.
[27]Venkateswarlu V,Manjunath K.Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles.J Control Release 2004;95:627-638.
[28]Huang L,Sullenger B,Juliano R.The role of carrier size in the pharmacodynamics of antisense and siRNA oligonucleotides.J Drug Target 2010;18:567-574.
[29]de Steenwinkel JE,van Vianen W,Marian T,et al.Targeted drug delivery to enhance eff i cacy and shorten treatment duration in disseminated mycobacterium avium infection in mice.J Antimicrob Chemother 2007;60:1064-1073.
[30]Van Hoesel Q,Steerenberg P,Crommelin D,et al.Reduced cardiotoxicity and nephrotoxicity with preservation of antitumor activity of doxorubicin entrapped in stable liposomes in the LOU/M Wsl rat.Cancer Res 1984;44:3698-3705.
[31]Zhang XK,Sun P,Bi R,et al.Targeted delivery of levofoxacinliposomes for the treatment of pulmonary inf l ammation. J Drug Target 2009;17:399-407.
[32]Bakker-Woudenberg IA,Marian T,Guo L,et al. Ciprof l oxacin in polyethylene glycol-coated liposomes: eff i cacy in rat models of acute or chronic pseudomonas aeruginosa infection.Antimicrob Agents Chemother 2002;46:2575-2581.
[33]Dupont B.Overview of the lipid formulations of amphotericin B.J Antimicrob Chemother 2002;49:31-36.
[34]Wong JP,Yang H,Blasetti KL,et al.Liposome delivery of ciprof l oxacin against intracellular Francisella tularensis infection.J Control Release 2003;92:265-273.
*Corresponding author.The School of Pharmaceutical Science,Shandong University,44 Wenhua Xi Road,Ji'nan,Shandong Province, China.Tel.:+86 0531 88382015;fax:+86 0531 88382548.
E-mail address:hgh2003@gmail.com(G.Huang).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2014.12.004
1818-0876/©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/3.0/).
 Asian Journal of Pharmacentical Sciences2015年3期
Asian Journal of Pharmacentical Sciences2015年3期
- Asian Journal of Pharmacentical Sciences的其它文章
- GUIDE FOR AUTHORS
- UPLC-MS/MS for the determination of azilsartan in beagle dog plasma and its applicationin a pharmacokinetics study
- Design and comparative in-vitro and in-vivo evaluation of starch-acrylate graft copolymer based salbutamol sulphate sustained release tablets
- Characterization of recrystallized itraconazole prepared by cooling and anti-solvent crystallization
- Enhanced bioavailability of rebamipide nanocrystal tablets:Formulation and in vitro/in vivo evaluation
- Chlorogenic acid loaded chitosan nanoparticles with sustained release property,retained antioxidant activity and enhanced bioavailability