
Enhanced bioavailability of rebamipide nanocrystal tablets:Formulation and in vitro/in vivo evaluation
2015-05-15 13:09:16YuGuoYongjunWangLuXu
Yu Guo,Yongjun Wang,Lu Xu
Shenyang Pharmaceutical University,No.103,Wenhua Road,Shenyang 110016,China
Enhanced bioavailability of rebamipide nanocrystal tablets:Formulation and in vitro/in vivo evaluation
Yu Guo,Yongjun Wang,Lu Xu*
Shenyang Pharmaceutical University,No.103,Wenhua Road,Shenyang 110016,China
ARTICLEINFO
Article history:
Received 24 June 2014
Received in revised form
30 August 2014
Accepted 2 September 2014
Available online 30 December 2014
Rebamipide
The purpose of this study was to formulate rebamipide nanocrystal tablets(REB-NTs)by wet-milling technique to enhance its dissolution rate and oral bioavailability.The formulation and preparation technology were screened by single factor tests with particle size and distribution as indicators.Rebamipide nanocrystals(REB-NSs)was then achieved by freeze-dry from the prepared nanosuspensions which were characterized by differential scanning calorimetry(DSC)and x-ray powder diffraction(XRD),while the vitro dissolution and the plasma drug concentration of the nanocrystal tablets were investigated.The results indicated that the prepared nanosuspensions got an average particle size of 286 nm, PI of 0.173 and the average Zeta potential of-18.2 mv.The average particle size of obtained REB-NSs’redispersibility was 278 nm,and the crystalline of REB-NSs was the same as the rebamipide bulk drug as shown by DSC and XRD.The drug dissolution rate of self-made nanocrystal tablets in different dissolutions was slightly faster than that from the reference tablets,REB-MTs(Mucosta®),while the Cmaxand AUC0-24of REB-NTs were 1 and 1.57 times higher than that of REB-MTs,which means the nanotechnology could signif i cantly improve the oral bioavailability of rebamipide.
©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/ licenses/by-nc-nd/4.0/).
1.Introduction
Rebamipide(REB)is a quinolone derivative drug,which is used to treat gastric and gastric mucosal lesions in acute gastritis and acute exacerbation of chronic gastritis[1,2].Because of its low solubility and low permeability,the bioavailability of rebamipide is under 10%in humans,thus rebamipide is classif i edintobiopharmaceuticsclassif i cationsystem IV(BCSIV)[3].Referred to solubility test of the literature[3], RBM was nearly indissolvable not only in polar but also in non-polar solvents.Shi YJ[4]prepared rebamipide nanosuspensions and improved its oral bioavailability.Nanosuspensions,also called nanocrystals,was regarded as an alternative and promising approach to settle these problems. It is a type of submicron colloidal dispersion system,wherein drug particles are distributed in water with a surfactant or polymer as a stabilizer through a self-assembly or broken preparation technology[5-7].As a potential technique,nanosuspension has been applied more and more widely to increase the solubility of poorly soluble drugs lately[8].The solubility was improved by reducing the drug particle size into the nano(sub-micron)range in the usage of nanocrystal technique.Inthisway,saturationsolubility(Cs)was increased,then dissolution rate(dc/dt)and bioavailability(F%) related to the formulation of poorly soluble drugs could be enhanced.At present,bothtop-downand bottom-upmethods are widely used in the f i eld of research in the new dosage form.The top-down wet milling technique can provide highly nanocrystalline with improved physical stability compared with the bottom-up micro-f l uidic precipitation method[9,10].
In this study,we adopt wet-milling method to prepare REBNTs.The goal of this research was to develop a formulation of REB-NTs for oral administration and to investigate the relative bioavailability in vivo using a rat model.A commercial REB product,Mucosta®,was used as a control for the dissolution and in vivo assay.
2.Materials and methods
2.1.MaterialsREBwas synthesized and donatedby Department of Medicinal Chemistry,Shenyang Pharmaceutical University.PVP K30, HPMC K4M and HPMC E5 were supplied by Anhui Shanhe Pharmaceutic Adjuvant Co.Ltd,Pluronic F68(poloxamer 188) was obtained from BASF(Germany).The commercial rebamipide tablets(brand name Mucosta®,100 mg/pill,Zhejiang dazhong zhiyao Co.Ltd,Batch No.121216R)as the reference product.All the other chemicals were of analytical grade or chromatographic grade.
Male SD rats weighing about 220 g(200-240 g)were obtained from the Experiment animal center of Shenyang Pharmaceutical University(Shenyang,China).Animal experiments were performed in compliance with the Guide for the Care and Use of Laboratory Animals and the declaration announced by the Animal Ethics Committee of Shenyang Pharmaceutical University.
2.2.Methods
2.2.1.Solubility determination of REB
The solubilities of REB in different pH aqueous solutions were determined.Excess Rebamipide was added to the buffer solutions,and all the mixtures wereshaken in an air bath shaker for 72 h at 37°C,then centrifuged at 3500 rpm for 10 min to remove supernatants which were appropriate diluted with the same buffer solutions before being analyzed by HPLC. HPLC condition:HITACHI 2000 HPLC system(HITACHI Pump L-2130,HITACHI UV Detector L-2420,HITACHI AutoSampler L-2200,Japan,Hitachi)with an Welch Ultimate®AQ-C18 (150 mm×4.6 mm,5 μm)(Welch,Shanghai,China);the mobile phase was acetonitrile-phosphate buffer solution mixture (17:83,v/v);the column temperature was maintained at 35°C and the wavelength was 254 nm;20 μl of sample was injected into HPLC system with the f l ow rate of 1 ml/min.Each sample was analyzed in triplicate.
2.2.2.Preparation of REB-NTs
The nanosuspensions were prepared by a bead mill(ESW-750, Shanghai Yile electromechanical Co.Ltd.,Shanghai)using wet-milling technique.Different types of stabilizers(F68, HPMC K4M,HPMC E5,PVP K30)were dissolved in 100 ml water with the concentrations of 5%(w/v),then 20 g of drug powder was dispersed in the aqueous stabilizer solution.The obtained suspensions were added in the milling bowl where the particles were broken into pieces by milling pearls(zirconium oxide,diameter 0.2 mm).The milling time was 30 min.In order to improve the stability of REB nanosuspensions,the obtained nanosuspensions were freeze-dried using FDU-1100 (EYELA,Tokyo Rikakikai Co.Ltd,Japan).A variety of freeze drying protectors including glucose,maltose,lactose,sucrose and mannitol were investigated with the concentrations of 3% (w/v).The freeze-drying process was as follows:f i rst,1 ml of the nanosuspensions were poured into a glass bottle with the diameter of 20 mm and placed at-80°C for 12 h,and subsequently lyophilized at a temperature of-40°C for 3 h,followed by a secondary drying phase of 16 h at-20°C,and then about 1 h at 10°C.The obtained freeze-dried powders were mixed with suitable adjuvants,then compressed into tablets by wet granulation.
2.2.3.Particle size and zeta potential
A Zetasizer Nano(Malvern Instruments,UK)was used to determine the mean particle size(z-average)and the polydispersity index(PI)which was regarded as a measurement of the width of size distribution.Before the measurement,the samplesweredilutedwithdistilledwatertoasuitablescattering intensity and re-dispersed by handshaking,then each sample was determined in triplicate with 13 runs and 60 s duration in each measurement at 25°C.Because of its ability of expressing the electric charge at the surface of the particles,the zeta potential(ZP)wascommonlyusedasameasurementofindicating thephysicalstabilityofcolloidalsystems.Inthisstudy,thesame Malvern Zetasizer was used to measure the ZP values by determiningtheparticleelectrophoreticmobilityoftheparticlesinan electrical f i eld,which was transformed to the zeta potential.
2.2.4.Differential scanning calorimetry(DSC)
DSC analysis was carried out using a differential scanning calorimeter(DSC 1 STARe,Mettler-Tolelo International Inc, Scotland).Samples(REB bulk drug,blank excipients,physical mixtures and REB-NSs)of about3 mgwere weighedaccurately and put in standard aluminum pans and sealed with a lid.The phase transition of sample was analyzed by differential scanning calorimetry at a heating rate of 10°C/min from 5°C to 350°C with a nitrogen purge of 20 ml/min.
2.2.5.X-ray powder diffraction(XRD)
Samples(REBbulkdrug,blankexcipients,physicalmixturesand REB-NSs)were evaluated with X-ray diffractometer(D/MAX-2400,Rigaku,Japan).Thediffractionpatternwasperformedina stepscanmodeloverananglearrangeof3°<2θ<45°,withastep size of 0.02°.
2.2.6.Dissolution tests for REB-NTs
Dissolution tests were performed according to Apparatus II (paddle)methodof JP X IV usingZRS-8G(Tianjin Tianda TianfaTechnology Co.Ltd.,Tianjin,China).Water with different concentration of SDS,pH 5.0 with different concentration of SDS,pH 1.2 HCl solution with 0.5%Tween 80,pH 6.0(1→4) phosphates-citric acid buffer solution and pH 6.8 PBS were used as the dissolution media respectively.The dissolution tests were carried out at the temperature of 37°C in 900 ml dissolution media,and the rotary speed of the paddles was set at 50 rpm.REB-NTs and REB-MTs were placed in the dissolution media,samples were withdrawn at 5,10,15,20,30,45 and 60 min,and immediately passed through a 0.45 μm millipore fi lter.The initial fi ltrate was discarded and the continuous fi ltrates was appropriately diluted.The mixtures were further analyzed by UV spectrophotometer(UV-1801,Beijing Rrayleigh Analytical Instrument company,China)at 326 nm.
2.2.7.Drug administration and blood plasma collection
The pharmacokinetics of the test preparations(REB-NTs)were compared with that of a reference formulation of Mucosta®tablets(REB-MTs).Test and reference samples were prepared by grinding with a pestle and mortar,then the powders were sieved through a 100-mesh sieve and suspended in a solution of sodium carboxymethyl cellulose 0.5%.Twelve Male SD rats were randomly assigned into two groups,then fasted for 12 h before drug administration and food was reoffered 4 h after dosing.The two solutions were administered orally to two groups of rats respectively at a dose of 10 mg/kg 200 μl blood samples were withdrawn from the posterior vena orbitalis at 0.08,0.25,0.5,1,1.5,2,2.5,3,4,6,8,10,12 and 24 h postdosing and placed into heparinized tubes.Blood samples were instantly centrifuged at 13,000 rpm for 10 min.Plasma was separated from samples and stored at-20°C until the time of analysis.
2.2.8.Sample preparation
Before analysis,the plasma samples were thawed at ambient temperature.100 μl plasma was added to a test-tube,then 50 μl internal standard solution and 50 μl water:methanol (50:50,v/v)were pipetted into the same tube,respectively. After a thorough vortex mixing for 5 min,mixtures were precipitated with 200 μl methanol,vortex-mixed vigorously for 5 min,and then the tube was centrifuged at 13,000 rpm for 10 min.Its supernatants(10 μl)were injected into the LC-MS/ MS system.
2.2.9.Drug concentration analysis in vivo
The LC-MS-MS method was set up to determine the drug concentrations in plasma.The assay was performed using Waters ACQUITY UPLC™/Tandem Quadrupole Detector system(Waters Crop.,Milford,MA).Liquid chromatographic separations were carried out by using a ACQUITY UPLC™BEH C18 column(50 mm×2.1 mm,1.7 μm,Waters Corp,Milford, MA,USA).The column and autosampler tray temperature were maintained at 35 and 4°C,respectively.The mobile phase consisting of methanol(A)and water containing 0.2% formic acid(B)was used at a f l ow rate 0.2 ml/min.The gradient elution was from 45%A to 60%A within 0.4 min and then held for 0.8 min,from 1.2 to 1.3 min,the percentage of A was increased to 70%and maintained about 1 min,then reduced to the initial condition within 0.2 min and balanced for 0.5 min.The total running time was 3 min and the sample injection volume was set at 10 μl.The UPLC system was connected with the mass spectrometer through an ESI interface and was operated in the positive ion detection mode. Nitrogen used for desolvation was 650 l h-1,cone gas was set at 50 l h-1.The capillary voltage was set at 2.85 KV,Source temperature 120°C,desolvation temperature 350°C.The compounds(REB and IS)were analyzed using multiple reaction monitoring(MRM)mode at m/z 371.3→216.3(cone voltages=28 v,collision energy=25 v)for REB,and m/z 278.3→58.04(cone voltages=12 v,collision energy=17 v)for venlafaxine(IS).Data acquisition was processed with Masslynx 4.1 software.
The calibration curves were linear(r2≥0.99)over the concentration range of 40-4000 ng/ml with the lower limit of quantif i cation(LLOQ)of 40 ng/ml with relative standard deviation(R.S.D.)lower than 10%.The extraction recoveries of REB were 94.8,89.1,and 93.2%at the concentrations of 80,500 and 3200 ng/ml,respectively.Mean recovery for the IS was 86.8%.The intra-day precisions,calculated from quality control(QC)samples at three concentration levels of 80,500,and 3200 ng/ml,was found to be below 6%.The inter-day precisions as determined from QC sampling was within 7%.
2.2.10.Statistical analysis
Pharmacokinetic analysis was carried out by means of a model independent approach.The maximum plasma concentration of REB(Cmax)and corresponding peak time(Tmax) were obtained by observation of the individual drug plasma concentration-time prof i les.The area under the curve to the last data point(AUC0-t)was calculated by the trapezoidal rule. The slopeof the terminal four pointsin plasmaconcentrationtime curve was conducted as the elimination rate constant (Ke).The elimination half-life(t1/2)of the preparation was calculated by 0.693/Ke.The relative bioavailability(F%)was calculated as follows:
F%=AUC0-t(test)/AUC0-t(reference)×100%
3.Results and discussion
3.1.Solubility determination of REB
As a water insoluble compound,it was necessary to investigate the determination of the solubility of REB,which was regarded as the background of selecting the dissolution medium.Table 1 showed the saturation solubility of REB bulk drug in different solution,including methanol,alcohol,pH 1.0 hydrochloric acid buffer solution,pH 4.5 acetate buffer solution,pH 5.0,5.5,5.8,6.0,6.8,7.4 phosphates buffer solution and pH 6.0(1→4)phosphate-citric acid buffer solution.The saturation solubility of REB in 0.1 mol/l HCl was extremely low being only 0.33 μg/ml.The solubility was increased with the increase of pH value.In this study,the saturation solubility of REB in water was 25 μg/ml,while the notable enhancement of the solubility was found to be in the pH value of 7.4 to be 376 μg/ml.And the results indicated that the sink condition for 100 mg tablets could be achieved in the dissolution medium pH 6.0(1→4)phosphates-citric acid buffer solution.

Table 1-Saturation solubility of REB in different pH media at 25°C.
3.2.Effect of wet-milling time
In our research,milling-time was investigated in the process of wet-milling.While the same stabilizer concentration (5%stabilizer,w/v)was added,mean particlesizes(Z-Average) of REB nanosuspensions but different milling times were displayed in Fig.1.With increasing milling times,the mean particle of REB was reduced.After 5 min of milling,the mean particle size was decreased obviously to be about 600 nm. However,when the milling time exceeded 30 min,the mean size of the particle was changed slightly.
3.3.Choice of the stabilizer and its concentration
Four stabilizers including F68,HPMC K4M,HPMC E5 and PVP K30 were chosen to be investigated.As can be seen in Figs.2 and 3,with constant milling time of 30 min,the mean particle sizesandpolydispersity index(PI)wereinf l uencednotonly by different stabilizers but also by the concentrations of stabilizer.The mean size of REB particles decreased to 326 nm stabilized by F68,358 nm stabilized by PVP K30,313 nm stabilized by HPMC K4M and 284 nm stabilized by HPMC E5 (Fig.2).HPMC E5 was the most effective stabilizer for REB with a lower PI of 0.178,followed by HPMC K4M.With different concentration levels of HPMC E5 as the stabilizer,the disparities of mean particle size and PI of drug particles were refl ected in Fig.3.Nanosuspensions prepared using 2%and 5% of HPMC E5 contributed the average diameter of 363 nm and 284 nm,correspondingly.As increasing the concentration of the stabilizer,i.e.,8%and 10%,the particle size remained almost the same.
3.4.Determination of particle size and zeta potential
The measurement of the mean particle size and zeta potential of suspensions represented a good estimation about formulation stability[11].The mean particle size and zeta potential of REB nanosuspensions were 286 nm and-18 mV,respectively.Even the existence of electrostatic stabilization,it could maintain a short time stability merely[12,13].In order to improve its'long-term stability,lyophilization was adopt to convertnanosuspensionsintopowdersforfurthermore investigation,i.e.,compressed into nanocrystal tablets.REBNSs freeze-dried in the presence of cryoprotectant possessed a good redispersibility with a mean particle size of 278 nm and PI of 0.180,a much smaller size of 344 nm(PI=0.280)without cryoprotectant.
3.5.DSC and XRD analysis
The REB bulk drug,freeze-dried powder,physical mixture of freeze-dried powder,and blank excipients of freeze-dried powder of all the materials were analyzed by DSC and XRD (Figs.4 and 5).As shown in Fig.4,the DSC curve of REB bulk drug exhibited a single sharp endothermic melting peak at 305°C,in accordance with literature[3],the endothermic peak of blankexcipientsdemonstrated at the temperature of 80and 153°C,respectively.In physical mixtures,the same endothermic peaks could be checked out simultaneously.The REBNSs showed a slightly shifted endothermic melting peak (300°C)compared to the bulk REB.However,the melting point of the bulk REB was higher than that of the REB-NSs and this phenomenon could be explain as particle size reduction [14,15].It could be deduced that there was no substantial crystalline change.As shown in Fig.5,it was supposed that almost no crystalline changes was detected in REB-NSs, because of the peaks in XRD of REB-NSs were in accordance with that in the physical mixture.In conclusion,the characteristic results conf i rmed that the crystalline state of REB had been unchanged after wet-milling and freeze-dry.
3.6.Dissolution tests
The in vitro drug dissolution test was carried out with different dissolution media.As mentioned in 3.1,pH 6.0 (1→4)phosphates-citric acid buffer solution was def i ned as sink condition for 900 ml dissolution medium and a f i xed dose of 100 mg REB,while the other media referred in the research except pH 6.8 phosphate buffer solution turned out to be oversaturation dissolution media.Almost 100%of the drug in REB-NTs was released within 45 min in pH 6.0(1→4) phosphates-citric acid buffer solution and pH 6.8 media, whereasthe dissolution rateof REB-MTswas slightlyslowerat the f i rst 20 min.The dissolution amount from REB-NTs in the oversaturation dissolution media were much higher than that from REB-MTs,especially in water with different concentration of SDS,in which the released drug from REB-NTs was about 1 time greater than that of REB-MTs(Fig.6).
According to the Noyes-Whitney and Ostwald-Freundlich equations,both particle size reduction and an augmentationofthesurfaceareawiththeenhancementof saturation solubility further resulted in an increase in the dissolution velocity[16,17].It was worth noting here that, within the f i rst 5 min,nearly no distinction was observed in the dissolution rate of two tablets in the same supersaturation media.That was conjectured that because the process of disintegration of the tablets cost some time to break into pieces,then drug dissolved from the fragments,and directly resulted in the lag-time phenomenon of drug concentration in those media.
3.7.In vivo evaluation
In our research,the concentrations of REB in samples obtained from plasmaof SD rats were determined by the method mentioned above.The UPLC-MS/MS method and extraction process were validated.The mean concentration-time curves for the two types of REB tablets are shown in Fig.7.The pharmacokinetic parameters including maximum peak concentration of the drug in plasma(Cmax),the time to reach maximum concentration(tmax),half life(t1/2),area under the curve(AUC)and elimination rate constant(ke)for both tablets were shown in Table 2.The higher Cmaxvalues of REB-NTs suggested that more drug was absorbed into blood after rapid dissolution in the gastrointestinal tract.The Cmaxand AUC0-24values of REB-NTs were approximately 1 and 1.57 times higher than that of REB-MTs,respectively.The relative bioavailability of REB nanocrystal tablets was 256.8%,which means the nanotechnology could signif i cantly improve the bioavailability of rebamipide.The effects could be interpreted that the higher drug concentration indicated drug molecules could be absorbed rapidly from gastrointestinal wall due to the notably elevated dissolution rate by decreasing particle size[18].
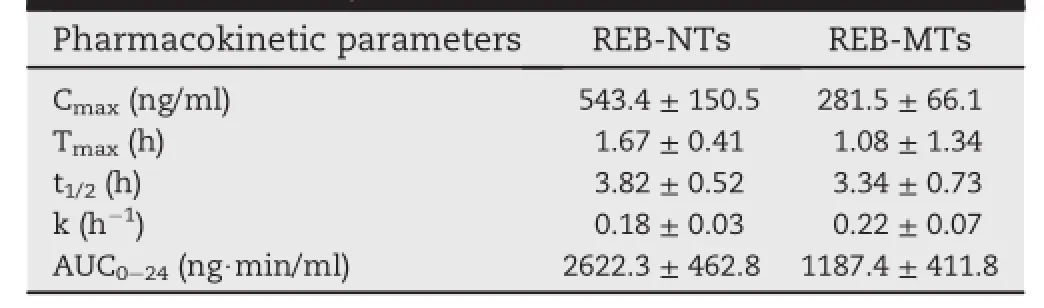
Table 2-Pharmacokinetic parameters following oral administration of REB-NTs and REB-MTs in a single dose of 10 mg/kg in SD rats(data were expressed as mean±SD,n=6).
4.Conclusions
The purpose of this study was to formulate and characterize REB-NTs to enhance the dissolution rate and oral bioavailability of this drug.The preparation method of nanosuspensions was supplied using wet-milling technique.HPMC E5 was considered to be an optimal stabilizer for obtaining the minimus particle size and PI.The X-ray powder diffraction (XRD)and differential scanning calorimetry(DSC)analysis indicated that no substantial crystalline change was detected in the nanocrystals compared with REB bulk drug.The in vivo study suggested that the Cmaxand AUC0-24values of REB-NTs in SD rats wereapproximately 1-foldand 1.57-foldhigherthan that of REB-MTs,respectively.In conclusion,nanocrystal drug delivery system was an effective strategy in enhancing the oral bioavailability of the insoluble drugs.
REFERENCES
[1]Huang BB,Li GF,Luo JH,et al.Permeabilities of rebamipide via rat intestinal membranes and its colon specif i c delivery using chitosan capsule as a carrier.World J Gastroenterol 2008;14:4928-4937.
[2]Arakawa T,Higuchi K,Fujiwara Y,et al.15th anniversary of rebamipide:looking ahead to the new mechanisms and new applications.Dig Dis Sci 2005;50:S3-11.
[3]Tung NT,Park CW,Oh T,et al.Formulation of solid dispersion of rebamipide evaluated in a rat model for improved bioavailability and eff i cacy.J Pharm Pharmacol 2011;63:1539-1547.
[4]Shi YJ,Zou MJ,An Y,et al.A potent preparation method combining neutralization with microf l uidization for rebamipide nanosuspensions and its in vivo evaluation.Drug Dev Ind Pharm 2012:1-9.Early online.
[5]Chingunpituk J.Nanosuspension technology for drug delivery.Walailak J Sci Technol 2007;4:139-153.
[6]Rabinow BE.Nanosuspensions in drug delivery.Nat Rev Drug Discov 2004;3:785-796.
[7]Tang XJ,Fu YH,Meng QH,et al.Evaluation of pluronic nanosuspensions loading a novel insoluble anticancer drug both in vitro and in vivo.Int J Pharm 2013;456:243-250.
[8]Ghosh I,Bose S,Vippagunta R,et al.Nanosuspension for improving the bioavailability of a poorly soluble drug and screening of stabilizing agents to inhibit crystal growth.Int J Pharm 2011;409:260-268.
[9]Tuomela A,Liu P,Puranen J,et al.Brinzolamide nanocrystal formulations for ophthalmic delivery: reduction of elevated intraocular pressure in vivo.Int J Pharm 2014;467:34-41.
[10]Ali HS,York P,Ali AM,et al.Hydrocortisone nanosuspensions for ophthalmic delivery:a comparative study between microf l uidic nanoprecipi-tation and wet milling.J Control Release 2011;149:175-181.
[11]Mu¨ller RH,Bohm BHL,Grau MJ.Nanosuspensionsformulations for poorly soluble drugs with poor bioavailability/Ist communication:production and properties.Pharm Ind 1999;61:74.
[12]Calcinari R.The zeta potential and its value in pharmaceutical technology.Farmaco Prat 1970;25:24-38.
[13]Carstensen JT,Stremming KP,Pothisiri P.Sedimentation kinetics of f l occulated suspensions.3.Effect of zetapotential.J Pharm Sci 1972;61:1999-2000.
[14]Xia DN,Quan P,Piao HZ,et al.Preparation of stable nitrendipine nanosuspensions using the precipitationultrasonication method for enhancement of dissolution and oral bioavailability.Eur J Pharm Sci 2010;40:325-334.
[15]Xu Y,Liu XY,Lian RY,et al.Enhanced dissolution and oral bioavailability of aripiprazole nanosuspensions prepared by nano-precipitation/homogenization based on acid-base neutralization.Int J Pharma 2012;438:287-295.
[16]Fu Q,Sun J,Zhang D,et al.Nimodipine nanocrystals for oral bioavailability improvement:preparation,characterization and pharmacokinetic studies.Colloids Surfaces B Biointerfaces 2013;109:161-166.
[17]Mu¨ller RH,Jacobs C,Kayser O.Nanosuspensions as particulate drug formulations in therapy:rationale for development and what we can expect for the future.Adv Drug Deliv Rev 2001;47:3-19.
[18]Hintz RJ,Johnson KC.The effect of particle size distribution on dissolution rate and oral absorption.Int J Pharm 1989;51:9-17.
*Corresponding author.Shenyang Pharmaceutical University,No.103,Wenhua Road,Shenyang 110016,China.Tel./fax:+86 24 23986293. E-mail address:xulu1974@hotmail.com(L.Xu).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2014.09.006
1818-0876/©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
Nanocrystal
Dissolution
Pharmacokinetics
 Asian Journal of Pharmacentical Sciences2015年3期
Asian Journal of Pharmacentical Sciences2015年3期
- Asian Journal of Pharmacentical Sciences的其它文章
- GUIDE FOR AUTHORS
- UPLC-MS/MS for the determination of azilsartan in beagle dog plasma and its applicationin a pharmacokinetics study
- Design and comparative in-vitro and in-vivo evaluation of starch-acrylate graft copolymer based salbutamol sulphate sustained release tablets
- Characterization of recrystallized itraconazole prepared by cooling and anti-solvent crystallization
- Liposomes for systematic delivery of vancomycin hydrochloride to decrease nephrotoxicity: Characterization and evaluation
- Chlorogenic acid loaded chitosan nanoparticles with sustained release property,retained antioxidant activity and enhanced bioavailability