
四氮杂大环含能化合物(TFFA)合成、表征与量子化学研究
2015-05-14 03:17:58王锡杰王伯周毕福强周彦水宁艳利
含能材料 2015年2期
王锡杰, 廉 鹏, 王伯周, 毕福强, 周彦水, 宁艳利
(西安近代化学研究所, 陕西 西安 710065)
1 引 言
呋咱是构建高能量密度化合物的有效单元[1],目前已合成出来的含能呋咱衍生物有几十种[2-4],其中性能较为突出的化合物是集呋咱、氧化呋咱环于一体的高能化合物3,4-双(3′-硝基呋咱-4′-基)呋咱氧化呋咱(DNTF)。目前以呋咱、氧化呋咱环于一体为基础结构引入偶氮基团合成新型大环含能材料,期望获得结构新颖、性能优异的新型含能化合物已成为含能材料研究领域的热点之一。2012年俄罗斯报道了一种十六环呋咱偶氮化合物[5]——四呋咱并[3,4-c:3′′,4′′-g:3′′′,4′′′-k:3′′′′′,4′′′′′-o]双氧化呋咱并[3′,4′-c:3′′′′,4′′′′-m] [1,2,9,10]四氮杂环十六辛烯(TFFA),熔点116~118 ℃,理论密度1.86 g·cm-3,生成焓1905 kJ·mol-1,爆速8775 m·s-1,爆压38.92 GPa,爆热为7015 kJ·kg-1。TFFA具有熔点低、能量密度较高、生成焓高、无氢高氮的特点,综合性能优异,有望作为熔铸炸药中的液相载体,也可作为固体推进剂氧化剂组分。
本研究参考文献[5],以3,4-双(4′-氨基呋咱-3′-基)氧化呋咱(DATF)为原料,经三氯异氰脲酸氧化合成了TFFA,并采用红外光谱、核磁共振﹑质谱和元素分析表征了结构; 针对文献[5]采用柱层析色谱柱分离法纯化TFFA产品时存在的操作复杂、污染较大的问题,本研究改进了纯化方法,采用有机溶剂乙腈、二氯甲烷与丙酮纯化TFFA产品; 探讨了三氯异氰脲酸氧化DATF的反应机理,推测了反应的微观过程,指导合成研究; 利用DSC、TG-DTG法研究了TFFA的热性能。运用Gaussian09程序的B3LYP方法,在6-31G(d,p)基组水平上对TFFA的结构进行了优化,得到了稳定的几何构型,在振动分析的基础上求得体系在不同温度下的热力学性质,得到了温度对热力学性能影响的关系式; 利用量子化学、VLW等方法,获得了TFFA的物化与爆轰性能,为应用研究提供基础数据。
2 实验部分
2.1 仪器与试剂
德国Bruker公司TENSOR 27型傅里叶变换红外光谱仪; 德国Elementar公司Vario EL Ⅲ型自动微量有机元素分析仪; 瑞士Bruker公司AV 500型(500MHz)超导核磁共振波谱仪; 日本岛津GC-MS-QP 2010 Plus型质谱仪; 北京泰克公司X-6熔点测定仪; 日本岛津GC-2010型高效液相色谱仪; 德国Netzsch公司DSC-204 HP高压差示扫描量热仪(DSC); 美国TA公司2950型热重-微商热重仪(TG-DTG)。
DATF为自制[6-8]; 乙腈,二氯甲烷,丙酮,均为分析纯,西陇化工股份有限公司; 三氯异氰脲酸,分析纯,常州格信化工有限公司。
2.2 实验过程
2.2.1 合成路线
以3,4-双(4′-氨基呋咱-3′-基)氧化呋咱(DATF)为原料,经三氯异氰脲酸氧化合成了TFFA。合成路线见Scheme 1:

Scheme1Synthetic route of TFFA
2.2.2 TFFA的合成
室温下将5.04 g(20 mmol)DATF加入到200 mL乙腈中,然后分批加入8.36 g(36 mmol)三氯异氰脲酸,室温反应2 h,过滤,用乙腈洗涤后得到黄色固体; 加入二氯甲烷溶解,过滤,蒸干有机溶剂,然后再用丙酮溶解,过滤、蒸干溶剂得黄色粉末2.6 g,收率为52.3%,纯度98.29%(高效液相色谱(HPLC)法,结果见图1),m.p.: 116~118 ℃。13C NMR(Acetone-d6, 125 MHz),δ: 103.821, 131.620, 133.711, 143.234, 160.296, 160.400; IR(KBr,ν/cm-1): 1649, 1605, 1578, 1550, 1513, 1422, 1126, 1046, 1029, 999, 968, 891, 804; MSm/z(%): 248 (13), 30 (100); Anal. calcd. for C12N16O8: C 29.03, N 45.16; found C 28.82, N 44.73。
图1TFFA高效液相色谱图
Fig.1High performance liquid chromatography(HPLC) spectrum of TFFA
2.2.3 TFFA热分析实验
差示扫描量热仪(DSC)操作条件: 试样量1.120 mg,试样皿为铝盘,气氛为流动氮气,流速为50.0 mL·min-1,压力为0.1 MPa,采用10.0 ℃·min-1的升温速率从室温升至400 ℃。
热重-微商热重仪(TG-DTG)操作条件: 试样量0.880 mg,试样皿为铝盘,气氛为流动氮气,流速为100.0 mL·min-1,采用10.0 ℃·min-1的升温速率从室温升至300 ℃。
3 结果与讨论
3.1 DATF氧化合成TFFA的反应机理探讨
DATF氨基氮原子有1对孤对电子,具有亲核性,进攻三氯异氰脲酸的Cl原子,生成氮鎓离子结构的中间体Ⅰ; 随后1分子中间体Ⅰ的氮鎓离子被另1分子中间体Ⅰ的氨基氮原子亲核进攻,脱去HCl得到大环中间体Ⅱ; 中间体Ⅱ进攻三氯异氰脲酸的Cl原子,最终生成双氮鎓离子结构中间体Ⅳ,然后发生分子内亲核反应,脱去HCl生成TFFA。推测其反应机理如Scheme 2所示。目前尚未进行实验验证,但根据文献[5]氧化剂与DATF的投料比计算,可知反应中存在二氯异氰尿酸。
3.2 谱学解析
3.2.1 红外光谱分析
图2为TFFA的红外光谱图。由图2可知,TFFA主要有以下几个强吸收峰: 1649,1605 cm-1属于氧化呋咱环上N—O键的伸缩振动,表明分子结构中存在氧化呋咱环; 1578 cm-1属于呋咱环上的C—N键的非对称伸缩振动,1422 cm-1属于呋咱环上的C—N键的对称伸缩振动以及伸缩振动引起的环的弯曲振动,1126 cm-1属于呋咱环上C—C键的面内弯曲振动或剪切振动,1046 cm-1属于呋咱环上N—O键的面内弯曲振动,表明分子结构中存在呋咱环; 804 cm-1属于偶氮基的面内弯曲振动。
3.2.2 TFFA质谱及裂解机理
图3为TFFA的有机质谱图,质量数最大的离子峰为m/z248,与TFFA分子量的一半相符,这是由于TFFA的分子量大且结构对称,在电子轰击源的轰击下,不易得到分子离子峰,可能在呋咱环与偶氮连接处裂解,产生与TFFA分子量一半符合的碎片离子峰。TFFA分子量为偶数,大部分碎片峰的m/z值也为偶数,判断该化合物含有偶数个氮原子,与分子中含有偶数个氮原子一致。根据谱图中低质量区的碎片m/z30、m/z54等可判断该试样结构中有呋咱环。通过碎片峰m/z232与质量数最大离子峰248相差16u,证明结构中有氧化呋咱环。

Scheme2Oxidation reaction mechanism of DATF
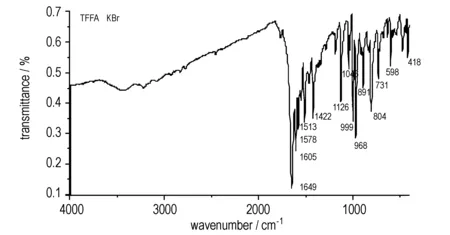
图2TFFA红外图光谱
Fig.2IR spectrum of TFFA
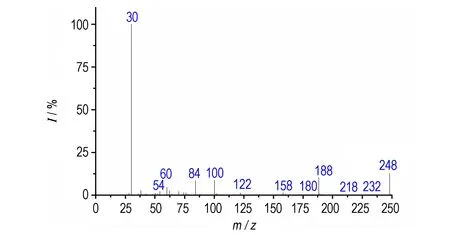
图3TFFA质谱图
Fig.3Mass spectrum of TFFA
3.2.3 核磁共振分析
图4为TFFA的13C NMR图谱,由图4可知,TFFA有6个峰,分别为δC103.821、δC131.620、δC133.711、δC143.234、δC160.296、δC160.400,其碳原子数及化学位移值与预定分子一致。

图4TFFA的13C NMR
Fig.413C NMR spectrum of TFFA
3.3 TFFA的热行为
常压(0.1 MPa)下TFFA的DSC曲线(升温速率β=10 ℃·min-1)如图5所示。由图5可见,TFFA有一个吸热峰和两个放热峰。吸热峰峰型尖锐,峰值温度为115.8 ℃,与TFFA熔点为116~118 ℃相吻合,表明样品经历吸热熔融为相变过程; 两个放热峰峰型较宽,峰值温度分别为215.2,293.8 ℃,峰型温度跨度较大,说明样品熔融为液体后气化分解。
常压下TFFA的TG-DTG曲线见图6。从图6中可知,TFFA受热第一阶段的质量损失大约为10.40%,可能是TFFA在升温熔融过程中的升华和发生“局部化学反应”[9]直接分解造成的; 第二阶段质量损失大约为74.41%,应为升温的过程中在自加热和自催化作用下样品较快分解的结果。当温度达到246.30 ℃分解基本完成,剩余残渣左右15.19%。

图5TFFA的DSC曲线
Fig.5DSC curve of TFFA

图6TFFA的TG-DTG曲线
Fig.6TG-DTG curves of TFFA
4 量子化学计算
4.1 计算方法和原理
由于B3LYP法较充分考虑电子相关,保持了从头算法等诸多优点,又较节省机时,且在6-31G(d,p)水平上求出的分子结构和性能接近于实验值,在含能材料领域已有广泛应用[10-12],本研究用Gaussian09[13]程序,对TFFA作DFT-B3LYP/6-31G(d,p)几何全优化计算,求得势能面上极小值,振动分析无虚频,进一步得到振动频率、IR谱及273~1000 K范围内的热力学性质。
4.2 结果与讨论
4.2.1 几何构型
TFFA在几何优化后的构型及原子编号见图7,键长、键角、二面角数据见表1,对几何优化后的构型进行振动频率计算,计算所得频率均为正值,表明所得构型为势能面上极小点,即相对稳定结构。
从图7和表1可以看出: 从偶氮基上四个氮原子组成的十六大环平面上看,TFFA的整个大环形成一个船型构型,两个氧化呋咱环分别处于船头位置,两个呋咱环处于船底位置,而另两个呋咱环则位于船底平面以下; 从侧面看,两个氧化三呋咱近似重合(见图8),这种结构使分子堆积更加紧密,结构更加稳定,从而表现出TFFA具有较高的密度。且环上的C—C和C—N 键长(1.305~1.439 Å)比标准的双键(1.22 Å)长,比标准的单键(1.46 Å)短,趋于平均化,所以每个呋咱环上形成一个小的共轭体系,增加了分子的稳定性。

图7B3LYP/6-31G(d,p)优化后的TFFA正面结构
Fig.7The frontal geometric configuration of TFFA optimized at B3LYP/6-31G(d,p)lever
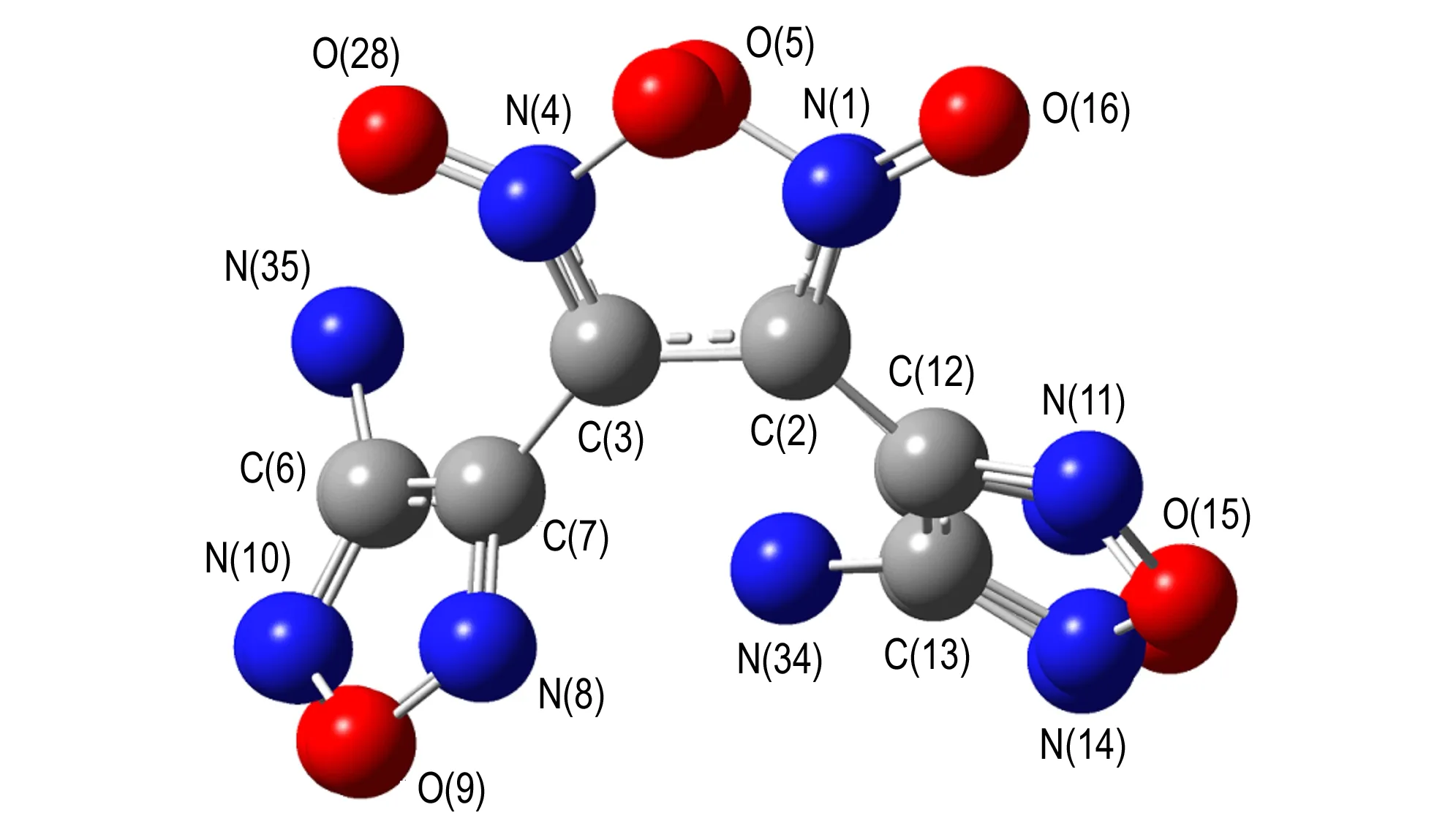
图8B3LYP/6-31G(d,p)优化后的TFFA侧面结构
Fig.8The side geometric configuration of TFFA optimized at B3LYP/6-31G(d,p) lever
表1B3LYP/6-31G(d,p)优化后的TFFA几何参数
Table1The geometric parameters of TFFA optimized at B3LYP/6-31G(d,p)level

bondlength/Åbondangle/(°)bonddihedralangle/(°)N(1)—C(2)1.345N(1)—C(2)—C(3)106.529N(1)—C(2)—C(3)—N(4)0.499N(1)—O(5)1.464C(2)—C(3)—N(4)111.385O(5)—N(4)—C(3)—C(2)0.207C(2)—C(3)1.429C(3)—N(4)—O(5)107.613O(5)—N(1)—C(2)—C(3)-0.925C(3)—N(4)1.310N(1)—O(5)—N(4)108.625C(3)—N(4)—O(5)—N(1)-0.786N(4)—O(5)1.355C(2)—C(3)—C(7)128.708C(2)—C(3)—C(7)—C(6)-114.076C(3)—C(7)1.472C(6)—C(7)—N(8)108.542C(2)—C(3)—C(7)—N(8)64.626C(6)—C(7)1.438C(7)—N(8)—O(9)105.478C(3)—C(7)—N(8)—O(9)178.986C(7)—N(8)1.305N(8)—O(9)—N(10)112.065C(6)—C(7)—N(8)—O(9)-2.062N(8)—O(9)1.372N(10)—C(6)—N(35)128.402C(7)—N(8)—O(9)—N(10)1.627C(2)—C(12)1.452C(7)—C(6)—N(35)122.730N(8)—O(9)—N(10)—C(6)-0.431C(6)—N(10)1.312C(6)—N(35)—N(36)124.966O(9)—N(10)—C(6)—N(35)-174.925O(9)—N(10)1.367O(5)—N(1)—O(16)118.767C(2)—C(12)—C(13)—N(14)176.668C(6)—N(35)1.410C(2)—C(12)—C(13)128.298C(12)—C(13)—N(14)—O(15)-0.809C(12)—C(13)1.443C(12)—C(13)—N(14)108.927C(13)—N(14)—O(15)—N(11)-0.532N(11)—C(12)1.311C(13)—N(14)—O(15)105.081N(14)—O(15)—N(11)—C(12)1.747C(13)—N(14)1.314C(12)—N(11)—O(15)105.339C(3)—C(2)—N(1)—O(16)-179.686N(11)—O(15)1.375N(11)—O(15)—N(14)112.560N(14)—O(15)1.360C(13)—N(34)—N(33)126.981C(13)—N(34)1.400N(33)—N(34)1.253
4.2.2 原子电荷
TFFA部分原子的净电荷列于表2(只给出对称轴一侧的数据)。由表2和图7可以看出: 呋咱环上与偶氮基相连的C(6)、C(13)原子带有较多的正电荷,这是由于偶氮基的吸电子作用和N原子的较强电负性所致,氧化呋咱上靠近配位氧一端的C(2)原子相比远离配位氧的C(3)原子所带正电荷更多,这是由于配位氧较强的电负性所致。
表2TFFA的原子电荷
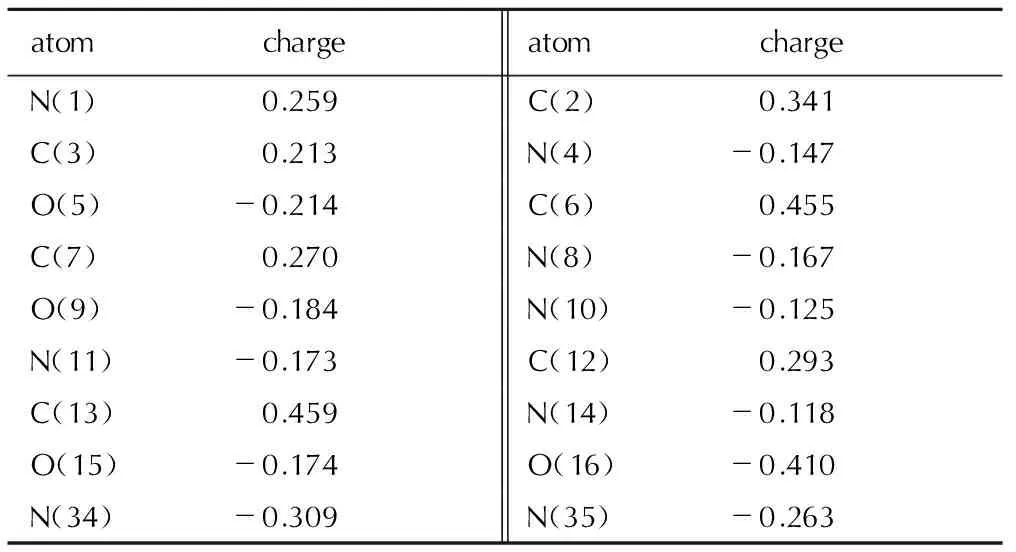
Table 2 Atomic charges of TFFA hartree
4.2.3 计算红外光谱
TFFA红外光谱计算结果(经过校正,校正系数为0.96)见图9。由图9可见,TFFA主要有以下几个强吸收峰: 1662,1654 cm-1属于氧化呋咱环上N—O键的伸缩振动; 1571 cm-1属于呋咱环上的C—N键的非对称伸缩振动和连接呋咱环的C—C键之间的伸缩振动以及伸缩振动引起的环的弯曲振动; 1461 cm-1属于呋咱环上的C—N键的对称伸缩振动以及伸缩振动引起的环的弯曲振动; 1329 cm-1属于呋咱环上C—C键的伸缩振动; 1157,1124 cm-1属于呋咱环上C—N键、C—C键的面内弯曲振动或剪切振动; 1032,1026 cm-1属于呋咱环上N—O键的面内弯曲振动或剪切振动; 821 cm-1属于偶氮基的面内弯曲振动。

图9TFFA的计算红外光谱(校正系数0.96)
Fig.9The calculated IR spectrum of TFFA with correction coefficient of 0.96
4.2.4 热力学性质
TFFA经B3LYP/6-31G(d,p)几何优化后求得的273~1000 K温度范围的标准热力学函数与温度关系曲线见图10。
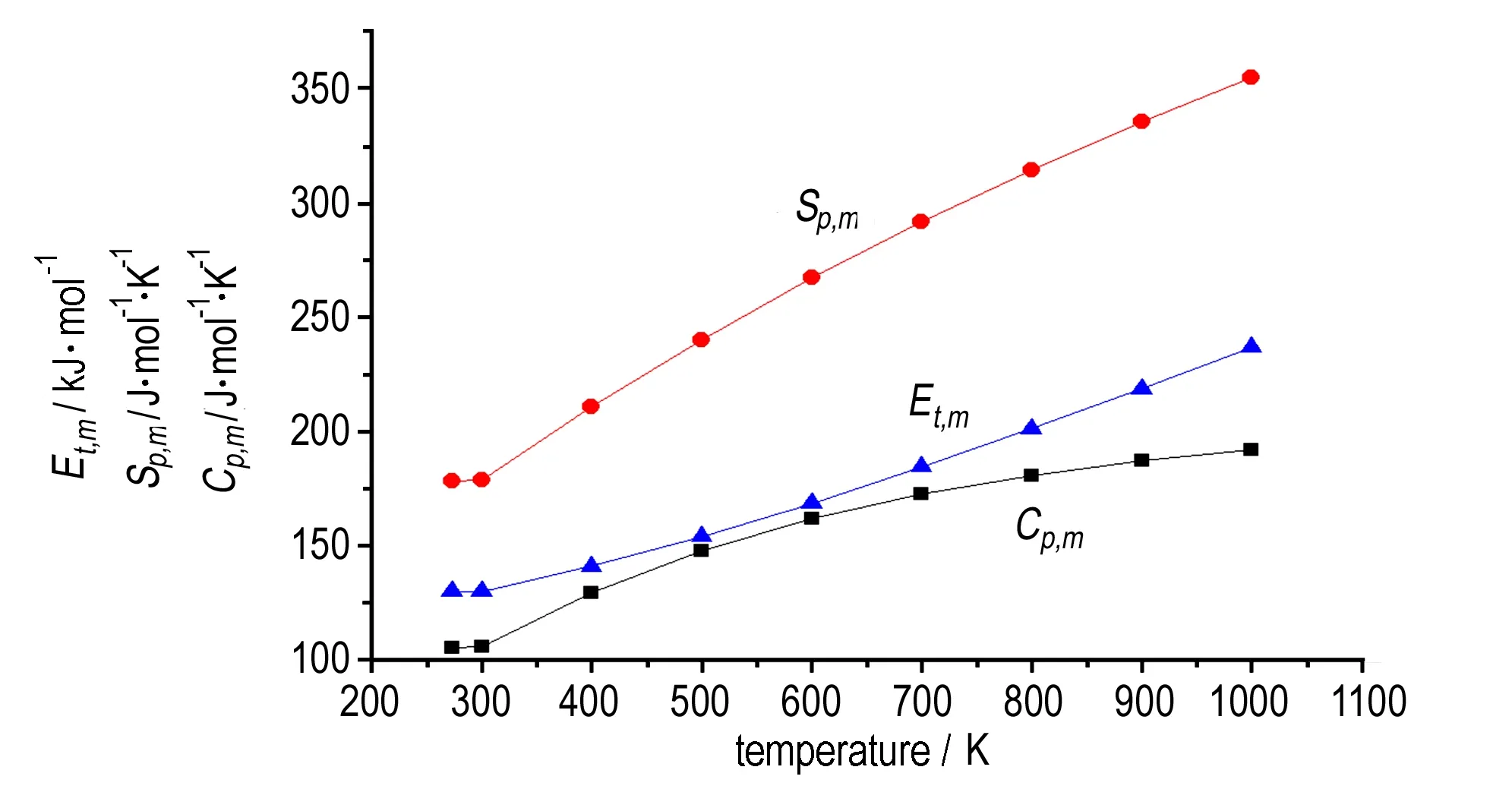
图10TFFA热力学性质与温度关系
Fig.10Relationships between thermodynamics properties of TFFA and temperature
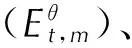
(1)
(2)
(3)
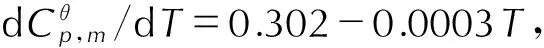
4.2.5 密度与生成焓
在B3LYP/6-31G(d,p)基础上,采用Monte-Carlo法[14]进行指定增加点密度的高精度分子体积计算,然后采用公式ρ=M/V计算得TFFA的密度为1.86 g·cm-3。
采用Politzer 等[15]推导的利用分子表面积和分子表面静电势计算固态物质升华焓,进而求得固相生成焓的方法(如式4~式7所示)计算得到化合物固相标准摩尔生成焓。其中,标准气相摩尔生成焓、分子表面静电势均通过B3LYP/6-31G(d,p)计算得到。
(4)
(5)
ΔHsub(298 K,kJ·mol-1)=0.000475A2+
(6)
ΔHf(solid,298 K)=ΔHf(gas,298 K)-ΔHsub(298 K)
(7)

(8)
(9)
经计算得TFFA生成焓为1905 kJ·mol-1。
4.2.6 爆轰性能
采用Kamlet-Jacobs公式[16]公式计算TFFA的爆速、爆压和爆热。对于CαHbOcNd炸药,其爆速和爆压可以用下述公式计算:
(10)
(11)
由于TFFA满足b/2≤c≤2a+b/2,因此上式中:
N=(b+2c+2d)/4M
(12)

(13)
(14)
式中,D为爆速,km·s-1;p为爆压,GPa;Q为爆热,kJ·kg-1;ρ0为炸药装药密度,g·cm-3;M为炸药分子量; ΔfHm为炸药标准摩尔生成焓,kJ·mol-1。
经计算得TFFA的爆速为8775 m·s-1,爆压为38.92 GPa,爆热为7015 kJ·kg-1。
5 结 论
(1) 以DATF和三氯异氰脲酸为原料,通过氧化反应获得新型含能化合物TFFA,改进纯化方法,使其纯度达98.29%,并采用核磁共振、红外、质谱以及元素分析等表征了结构。
(2) 探讨了以三氯异氰脲酸为氧化剂氧化DATF的反应机理,推测了反应的微观过程; 利用DSC、TG-DTG等研究了TFFA的热行为,其热分解峰温为215.2 ℃,表明热稳定性较好。
(3) 量子化学计算表明: TFFA的整个大环形成一个船式构型,偶氮基的吸电子作用和N原子的较强电负性,使得呋咱环上与偶氮基相连的C(6)、C(13)原子带有较多的正电荷,从而增加了整个分子的稳定性。计算了TFFA的爆轰性能,其密度1.86 g·cm-3,生成焓1905 kJ·mol-1,爆速8775 m·s-1,爆压38.92 GPa,爆热为7015 kJ·kg-1。
参考文献:
[1] Coburn M D. Picryiamino-substituted heterocycles[J].JHeterocyclChem, 1968, 5: 83-87.
[2] Sheremeteev A B, Kulagina V O. Furazan derivatives: High energetic materials from diaminofurazan[C]∥22th International Pyrotechnics Seminar Colorado: International Pyrotechnics Society, 1996, 377-378.
[3] 李加荣. 呋咱系列含能材料的研究进展[J]. 火炸药学报,1998,21(3): 56-59.
LI Jia-rong. Research development of furazanenergetic materials[J].ChineseJournalofExplosivesandPropellants, 1998, 21(3): 56-59.
[4] 霍欢, 王伯周, 王锡杰, 等. 呋咱含能化合物的合成及其衍生物反应研究进展[J]. 化学推进剂与高分子材料, 2013, 11(3): 15-22.
HUO Huan, WANG Bo-zhou, WANG Xi-jie, et al. Research progress in synthesis of furazan energeticcompounds and reaction of their derivatives[J].Chemicalpropellants&polymericmaterials, 2013, 11(3):15-22.
[5] David E C, Damon AP. The synthesis and characterization of a new furazan heterocyclic system[J].Synletter, 2012, 23: 2126-2128.
[6] 周彦水, 王伯周, 李建康, 等. 3,4-双(4′-硝基呋咱-3′-基)氧化呋种合成、表征与性能研究[J]. 化学学报, 2011, 69(14): 1673-1680.
ZHOU Yan-shui, WANG Bo-zhou, LI Jian-kang, et al. Study on synthesis, characterization and properties of 3,4-bis(4′-nitrofurazano-3′-yl)furoxan[J].ActaChimSinica, 2011, 69(14): 1673-1680.
[7] 胡焕性, 张志忠, 王亲会, 等. 高能量密度材料3,4-二硝基呋咱基氧化呋咱的性能及应用研究[J]. 兵工学报, 2004, 25(2): 155-158.
HU Huan-xing, ZHANG Zhi-zhong, WANG Qin-hui, et al. A study on the properties and application of high energy density material DNTF[J].ActaArmamentarii, 2004, 25(2): 155-158.
[8] 周彦水, 李建康, 黄新萍, 等. 3,4-双(4′-氨基呋咱-3′)氧化呋咱的合成及性能[J]. 火炸药学报,2007, 30(1): 54-56.
ZHOU Yan-shui, LI Jian-kang, HUANG Xin-ping, et al. Synthesis and properties of 3,4-bis (4′-aminofurazano-3′)furoxan[J].ChineseJournalofExplosives&Propellants, 2007, 30(1): 54-56.
[9] 刘子如. 含能材料热分析[M]. 北京: 国防工业出版社,2008.
LIU Zi-ru. Thermal Analyses for Energetic Materials[M]. Beijing: National Defense Industry Press, 2008.
[10] 来蔚鹏, 廉鹏, 王伯周, 等. 二硝基吡唑并吡唑性能的量子化学研究[J]. 计算机与应用化学, 2007, 24(8): 1025-1028.
LAI Wei-peng, LIAN Peng, WANG Bo-zhou, et al. Study of the properties of 3,6-dinitropyrazolo[4,3-c]pyrazoles by quantum chemistry[J].ComputersandAppliedChemistry, 2007, 24(8): 1025-1028.
[11] 梁晓琴. 四唑衍生物结构及性质的理论研究[J],四川师范大学学报(自然科学版),2008,31(2): 219-223.
LIANG Xiao-qin.Theoretical study on geometries and properties of tetrazole derivatives[J].JournalofSichuanNormalUniversity(NaturalScience), 2008, 31(2): 219-223.
[12] 马海霞,肖鹤鸣,宋纪蓉,等. 1,2,4-三唑-5-酮结构和性质的理论研究[J]. 含能材料, 2005, 13(3): 166-168.
MA Hai-xia, XIAO He-ming, SONG Ji-rong, et al. Theoretical study on the structure and properties of 1,2,4-triazol-5-one[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2005, 13(3): 166-168.
[13] Frisch M J, Trucks G W, Schlegel H B, et. al. Gaussian 09[CP], Gaussian, Inc, Wallingford CT, 2009.
[14] Ming W W, kenneth B W, Michael J F. Ab initio calculation of milar volumes: comparison with experiment and use in solvation models[J].JComputerChem, 1995, 16(3): 385-394.
[15] Politzer P, Murray J S, Grice M E, et al. Calculation of heats of sublimation and solid phase heats of formation[J].MolecularPhysics, 1997, 91(5): 923-928.
[16] Kamlet M J, Jacobs S J. Chemistry of detonationsI. A simple method for calculating detonation properties of CHNO explosives[J].JournalofChemicalPhysics, 1968, 48(1): 23-35.
猜你喜欢
兵器装备工程学报(2022年12期)2023-01-06 04:24:04
当代水产(2021年10期)2022-01-12 06:20:40
成都大学学报(自然科学版)(2021年1期)2021-05-22 01:31:18
火工品(2019年3期)2019-09-02 05:48:22
火炸药学报(2018年1期)2018-04-19 02:42:45
食品与机械(2017年5期)2017-07-05 13:24:36
环境科技(2016年4期)2016-11-08 12:18:58
中国粮油学报(2016年1期)2016-02-06 02:17:09
化工进展(2015年6期)2015-11-13 00:26:50
火炸药学报(2014年3期)2014-03-20 13:17:38
