
硬皮病伴发肺间质病变小鼠模型的建立及发病机制研究
2015-05-07 00:40曾海英
复旦学报(医学版) 2015年4期
张 立 李 正 曾海英 王 强
(1复旦大学附属中山医院皮肤科,2实验研究中心,3病理科 上海 200032)
硬皮病(systemic scleroderma,Ssc)是以局限性或弥漫性皮肤和内脏器官结缔组织的纤维化或硬化,最后发生萎缩为特点的疾病。是一种系统性或全身性自身免疫病,与系统性红斑狼疮和皮肌炎/多发性肌炎等都归类于自身免疫病范畴。近年来发现,硬皮病伴发肺间质病变(interstitial lung disease,ILD)发生率高达25%~90%[1],且是Ssc致死的首要原因。然而,目前硬皮病伴发肺间质病变(Ssc-ILD)发病机制仍不清楚,值得深入研究。本课题组通过不断调整博来霉素(bleomycin,BLM)浓度来摸索建立Ssc-ILD小鼠模型的适宜条件,最终以C3H/He小鼠局部(背部皮肤)皮下注射0.4 mg/mL BLM成功建模,小鼠肺部可见典型的炎症和纤维化。我们通过HE和Masson染色明确模型的可靠性,并通过检测羟脯氨酸了解纤维化程度及免疫组化检测CD8+T细胞、CD68+巨噬细胞、TGF-β1、IL-23、collagen-1、MMP-9和α-SMA 来初步探讨Ssc-ILD的发病机制。
材料和方法
实验动物 8只SPF级C3H/He小鼠由上海复旦大学附属中山医院动物中心提供,雌性,6周龄,体质量(20±2)g,于清洁级动物实验室饲养。饲养1周后进行实验。每只小鼠背部中央区域去毛使皮肤裸露,便于注射和观察。随机等分为两组,分别为造模组和对照组。
BLM局部皮下注射建模 盐酸博来霉素粉剂(日本化学株式会社,规格15 mg/支)用PBS缓冲液(pH=7.4)配制成浓度为3 mg/mL溶液,置于-20℃冰箱保存,临用前稀释成浓度为0.4 mg/mL的BLM。造模组在每天相同时间用0.4 mg/mL BLM背部皮肤皮下注射0.1 mL;对照组于相同部位皮下注射0.1 mL PBS,连续注射28天。
动物处理 观察实验过程中小鼠背部皮肤变化及被毛生长情况、活动度及健康状况。于第29天处死小鼠取标本。首先取皮,于注射部位处取1 cm×1 cm皮肤标本,一部分置于液氮中待测羟脯氨酸,另一部分浸于4%中性甲醛溶液中,待测病理和免疫组化;接着取肺,左叶存于液氮中,右叶浸于4%中性甲醛溶液中。
组织病理学观察 皮肤及肺组织标本予以4%中性甲醛溶液固定,脱水,石蜡包埋,切片(厚度3 μm),行常规HE染色及Masson染色。HE染色切片用Leica病理图像分析系统测定皮肤真皮厚度[2],每张切片随机取5个部位检测真皮厚度(即表皮与皮下组织之间的真皮垂直距离),取均值进行统计,比较造模组与对照组的差异。Masson染色切片用Leica图片定量分析系统测定其组织化学染色图像[3],每张切片随机取3处视野(×200),以蓝色为阳性,先选中视野范围内所有组织(除去空白部分)记录组织总面积,再选中蓝色部分记录胶原面积,计算胶原面积百分比(胶原面积百分比=胶原面积/组织总面积)。
胶原含量分析 取大小约0.3 cm×0.3 cm的皮肤和肺组织,加生理盐水用匀浆器研磨成10%的匀浆。用羟脯氨酸试剂盒(南京建成生物工程研究所)进行检测,加设标准品管,与待测样品管一同水浴消化,先后加Ⅰ、Ⅱ、Ⅲ 液,2000×g离心10 min。取上清液在分光光度计550 nm处(1 cm光径)空白管调零后检测各标本吸光度。各管羟脯氨酸含量的计算公式:羟脯氨酸含量=(测定D值-空白D值)/(标准D值-空白D值)×标准品浓度÷样本含量/所加匀浆介质。根据羟脯氨酸在胶原蛋白中占13.4%换算成胶原蛋白含量。
免疫组化染色 皮肤和肺组织经4%中性甲醛溶液固定、石蜡包埋、切片,CD8+、CD68+、TGF-β1、IL-23、collagen-1、MMP-9、α-SMA 抗体(抗体工作浓度为1∶100,美国Abcam公司)孵育,用二步法EnVision(ChemMafeTMEnVision+/HRP),DAB显色[二步法抗兔/鼠通用型免疫组化检测试剂盒,基因科技(上海)有限公司],进行免疫组化染色。在组织细胞膜或细胞质中出现棕黄色颗粒的为阳性,在200倍光学显微镜下随机选取3个视野,用Image Pro-Plus 6.0分析软件定量分析。
统计学分析 采用SPSS 20.0统计软件进行t检验。P<0.05为差异有统计学意义。
结 果
小鼠形态观察 造模组:背部皮肤注射部位出现皮肤显著增厚、硬化,弹性变差,注射点有破溃和结痂现象,毛发未恢复生长,小鼠体型变小;对照组:注射部位皮肤无明显变化,弹性较好,注射点无结痂,毛发恢复生长,小鼠体型无明显变化(图1)。

图1 小鼠大体形态变化Fig1 The general changes of mice
皮肤组织病理学观察 造模组小鼠相比对照组HE染色结果(图2A)显示真皮层显著增厚[图2D,(407.78±15.20)μmvs.(125.16±7.04)μm,P<0.01],胶原纤维束数量增多,形状增粗,排列紧密,向深部扩展至部分替代皮下脂肪组织;在真皮深部看到典型的血管壁增厚纤维化(图2C)。对照组小鼠皮肤组织无明显病理改变。
皮肤纤维化程度 造模组和对照组小鼠皮肤胶原面积百分比分别为75.23%±0.98%和13.84%±3.87%(图2B),皮肤胶原含量分别为(450.00±37.64)μg/g和(193.12±13.21)μg/g,造模组均高于对照组,差异均有统计学意义(P<0.01,图2E、图3)。同时发现,胶原以Ⅰ型胶原为主,并且造模组与对照组相比含量显著增多(图4A,63.45±5.90 vs.6.39±0.78,P<0.01)。
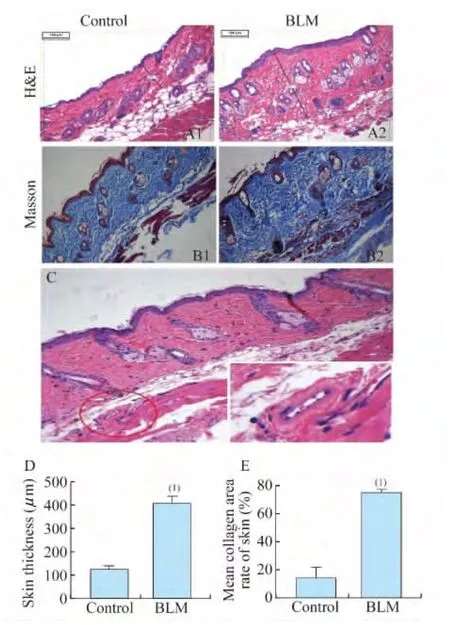
图2 小鼠皮肤组织学变化Fig2 Histological changes in the skins of mice
A:HE staining(×200);B:Masson′s staining(×200);C:Thickened wall of small blood vessel in dermis(×200);D:Skin thickness;E:Collanen area rate.vs.Control group,(1)P<0.01.

图3 小鼠皮肤和肺胶原含量比较Fig3 Comparison of content of collagen in skin and lung of mice
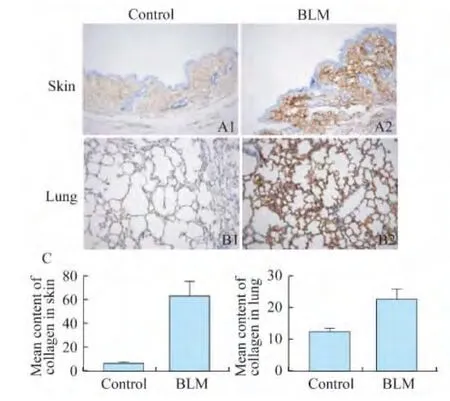
图4 小鼠皮肤和肺的Ⅰ型胶原含量比较Fig4 Comparison of collagenⅠin skin and lung fo mice
组织病理学观察 造模组小鼠肺组织中可见肺泡间隔增宽伴有炎性细胞渗出及浸润,肺泡结构破坏;对照组小鼠肺泡结构正常(图5A)。
肺部炎症程度 造模组和对照组小鼠肺部组织炎性细胞渗出主要为CD8+T细胞(图6A,40.37±1.07 vs.10.36±0.66,)和CD68+巨噬细胞(图6B,32.06±0.43 vs.5.83±0.31),造模组均显著高于对照组;造模组细胞因子IL-23也显著高于对照组(图6C,30.36±1.52 vs.6.87±0.41,P<0.01)。

图5 小鼠肺部组织学变化Fig5 Histological changes in the lungs of mice
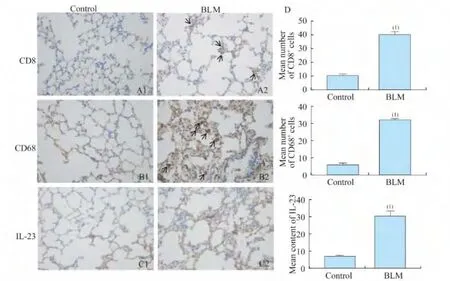
图6 小鼠肺部组织炎症变化Fig6 Changes of inflammation in the lungs of mice
纤维化程度 造模组和对照组小鼠肺组织胶原面积百分比分别为47.38%±5.01%和7.93%±3.97%(P<0.01),两组肺组织胶原含量分别为(314.93±23.96)μg/g和(142.62±9.36)μg/g,造模组均高于对照组,差异均有统计学意义(图5C,P<0.01)。造模组的Ⅰ型胶原表达显著增高(图4B,22.79±1.60 vs.12.58±0.51,P<0.01);TGF-β1表达显著增多(图7A,28.58±2.29 vs.5.17±0.74,P<0.01);MMP-9表达显著增多(图 7B,53.59±3.50 vs.5.27±1.8,P<0.01);标记肌成纤维细胞的α-SMA表达显著增高(图7C,50.64±1.19 vs.4.99±1.07,P<0.01)。
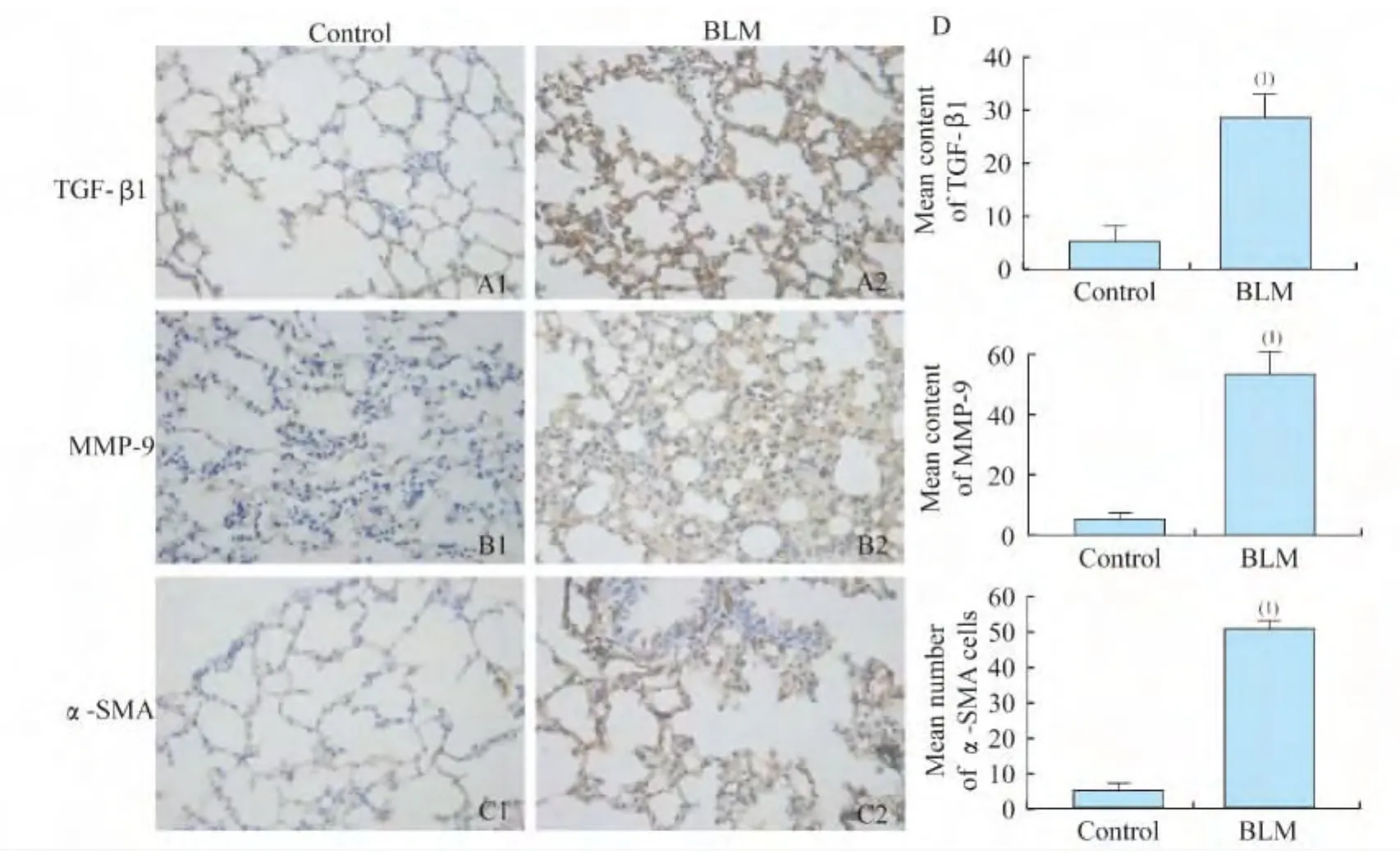
图7 小鼠肺组织纤维化指标变化Fig7 Changes of fibrosis in the lungs of mice
讨 论
ILD是增加Ssc等结缔组织病住院率和死亡率的首要原因,因此通过建立Ssc-ILD动物模型来探究其发病机制并寻找新的靶点,可为防治该组疾病提供新的思路。自Yamamoto等[4]成功诱导小鼠皮肤硬化以来,国内外众多学者尝试建立Ssc动物模型。文献报道,C3H/He等小鼠是对BLM十分敏感的动物品系[5]。BLM是一种广泛运用于肿瘤治疗的化疗药物,其不良反应包括肺纤维化或硬皮病样改变[6]。本研究前期发现,给予浓度0.1 mg/mL BLM连续皮下注射28天,小鼠皮肤出现轻度增厚,但肺部并没有炎性改变(图8A);而当调整浓度至0.2 mg/mL时,小鼠皮肤增厚明显,而肺泡壁轻度增宽但不明显(图8B);最终当浓度调整至0.4 mg/mL时,出现典型的皮肤硬化和肺部组织ILD改变,提示BLM的致肺部纤维化作用是呈浓度依赖性的,对临床化疗中用BLM的浓度有一定的提示意义。
本课题组在国内首次成功建立Ssc-ILD动物模型:以浓度为0.4 mg/mL BLM对C3H/He小鼠进行连续28天局部皮下注射,成功建立Ssc-ILD模型;小鼠不仅皮肤产生与Ssc相似的组织病理学改变,并且肺部组织也出现显著炎症和纤维化。在此基础上,本课题组初步探讨Ssc-ILD的发病机制。
炎症和纤维化是Ssc的特征性改变,由炎性细胞和细胞因子共同作用,导致成纤维细胞的活化和细胞外基质(extracellular matrix,ECM)沉积[7]。其中,转化生长因子-β(TGF-β)参与纤维化的全过程,在Ssc发病机制中起关键作用[8];尤其是TGF-β1,能促进成纤维细胞增殖分化为表达α-SMA的肌成纤维细胞,并分泌大量的ECM,启动纤维化过程[9-10]。Yamamoto 等[11]研究发现抗TGF-β治疗后,α-SMA表达显著减少,进一步肯定了TGF-β对成纤维细胞的刺激作用。有学者发现TGF-β致肺纤维化是通过下调成纤维细胞和单核细胞中的小凹蛋白-1(caveolin-1);因为Ssc患者成纤维细胞和单核细胞释放TGF-β增多使caveolin-1表达减少,从而抑制Smad3磷酸化同时增加TGF-β配体的内吞和降解,最终导致纤维化[12-13]。本研究结果表明,皮肤真皮显著增厚,胶原纤维堆积,小血管壁增厚纤维化;其中,TGF-β-1、collagen1、α-SMA 在造模组中显著增多,提示TGF-β1参与Ssc皮肤硬化过程,且可能是通过α-SMA导致真皮层collagen-1增多。
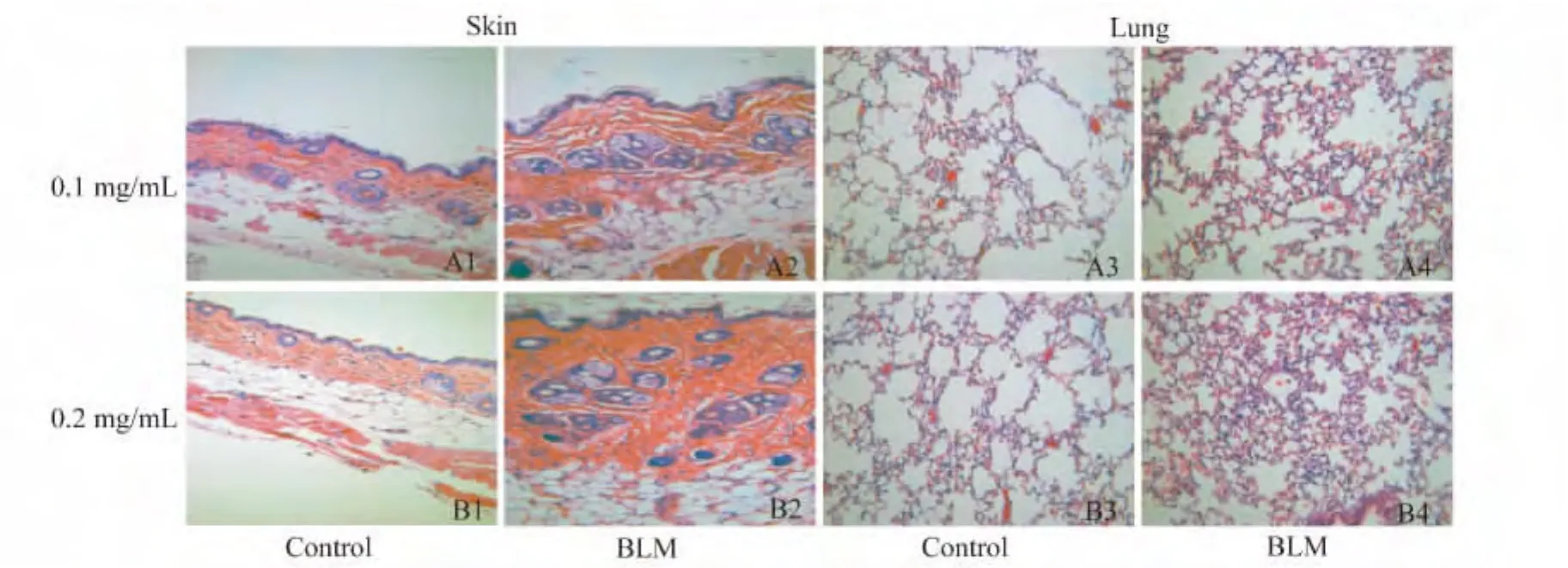
图8 0.1和0.2mg/mL BLM注射小鼠皮肤和肺变化(×200)Fig8 Changes of skin and lung after injection of BLM at concentration of 0.1and 0.2mg/mL(×200)
在Ssc-ILD中,成纤维细胞是肺纤维化启动和维持的主要细胞类型,并通过TGF-β介导,释放大量Ⅰ型胶原蛋白[14]。TGF-β1是炎症进展为纤维化的重要中间介质[15]。Ssc-ILD引起肺部炎症微环境发生变化,TGF-β、单核细胞趋化因子-1、巨噬细胞炎性蛋白等上调,导致淋巴细胞聚集和胶原纤维的沉积[16]。炎性细胞如T细胞、巨噬细胞等参与Ssc的发病进程,产生大量细胞因子导致ECM增多和血管损伤[17]。进一步研究明确是CD8+T淋巴细胞[18]和 CD68+巨噬细胞[19]参与发病过程。最新的基因芯片技术表明巨噬细胞的聚集活化和TGF-β基因上调与Ssc-ILD中肺纤维化进展密切相关[20]。目前认为TGF-β主要通过Samd和非Smad通路引起纤维化的发生,而近期亦有研究表明其作用同时与 Toll样受体4(TLR-4)有关。TLR-4能识别脂多糖(lipopolysaccharide,LPS),增加成纤维细胞对 TGF-β1的敏感性,引起由 TGF-β1介导的前Ⅰ型胶原α1链(COL1A1)mRNA和Ⅰ型胶原蛋白表达增多。在TLR-4基因敲除鼠中,α-SMA表达显著减少。TLR-4对正常成纤维细胞的刺激作用参与ECM重建和组织修复,协同增加TGF-β1调节的纤维化进程[21]。
MMP-9是近年来发现与肺纤维化相关的金属基质蛋白酶。纤维化早期病变的发生是由炎性反应(iNOS和 COX-2)、凋亡(p53和caspase-3)以及基质损伤(MMP-9、MMP-2和 COL1A1)引起[22]。在肺纤维化中,Thy-1阴性的成纤维细胞内 MMP和TGF-β1增多[23]。生长因子如 TGF-β在基因 水 平调控 MMP[24]。MMP-9在 BLM 引起的肺纤维化的支气管肺泡灌洗液中增高,而用 MMP抑制剂batimastat后能改善肺纤维化[25],提示 MMP-9参与肺纤维化进程。在硬皮病肺动脉高压中,MMP-9和血管内皮生长因子含量显著增多,其中 MMP-9通过胶原重建而刺激血管生成同时也控制血管过度形成[26]。
IL-23参与硬斑病(局限性硬皮病)发病。IL-17是许多炎症和自身免疫疾病的重要炎性因子,而IL-23是刺激IL-17合成的主要细胞因子[27]。
本实验结果表明,在模型组小鼠肺组织中,TGF-β1显著增多伴随着与纤维化密切相关的α-SMA、collagen-1、MMP-9 升高,而与炎症相关的CD8+T细胞、CD68+巨噬细胞以及炎性因子IL-23显著增多,上述观察性的现象和最新相关文献报道提示Ssc-ILD的发生极可能是通过上调TGF-β1,导致肌成纤维细胞活化和增生,由巨噬细胞提呈和CD8+T细胞协助,产生IL-23和TGF-β1,从而引起MMP-9增多,最终致ECM 增多,形成肺纤维化。目前,我们在动物模型上发现的现象仅初步探索了可能参与Ssc-ILD的因素,对于其发病机制还需结合体内体外试验作进一步深入研究。
[1]Winstone TA,Assayag D,Wilcox PG,et al.Predictors of mortality and progression in scleroderma-associated interstitial lung disease:a systematic review[J].Chest,2014,146(2):422-436.
[2]Kajii M,Suzuki C,Kashihara J,et al.Prevention of excessive collagen accumulation by human intravenous immunoglobulin treatment in a murine model of bleomycin-induced scleroderma[J].Clin Exp Immunol,2011,163(2):235-241.
[3]Zhang L,Fu XH,Yu Y,et al.Treatment with CA-074 Me,a Cathepsin B inhibitor,reduces lung interstitial inflammation and fibrosis in a rat model of polymyositis[J].Lab Invest,2015,95(1):65-77.
[4]Yamamoto T,Takagawa S,Katayama I,et al.Animal Model of Sclerotic Skin.I:Local Injections of Bleomycin Induce Sclerotic Skin Mimicking Scleroderma[J].J Invest Dermatol,1999,112(4):456-462.
[5]Oi M,Yamamoto T,Nishioka K.Increased expression of TGF-beta1 in the sclerotic skin in bleomycin-′susceptible′mouse strains[J].J Med Dent Sci,2004,51(1):7-17.
[6]Burger RM.Cleavage of Nucleic Acids by Bleomycin[J].Chem Rev,1998,98(3):1153-1170.
[7]Bhattacharyya S,Wei J,Varga J.Understanding fibrosis in systemic sclerosis:shifting paradigms,emerging opportunities[J].Nat Rev Rheumatol,2012,8(1):42-54.
[8]Varga J,Pasche B.Transforming growth factor beta as a therapeutic target in systemic sclerosis[J].Nat Rev Rheumatol,2009,5(4):200-206.
[9]Ihn H.Scleroderma,fibroblasts,signaling,and excessive extracellular matrix[J].Curr Rheumatol Rep,2005,7(2):156-162.
[10]Hinz B,Phan SH, Thannickal VJ,et al. The myofibroblast:one function,multiple origins[J].Am J Pathol,2007,170(6):1807-1816.
[11]Yamamoto T,Nishioka K.Animal model of sclerotic skin.V:Increased expression of alpha-smooth muscle actin in fibroblastic cells in bleomycin-induced scleroderma[J].Clin Immunol,2002,102(1):77-83.
[12]Trojanowska M.Noncanonical transforming growth factor beta signaling in scleroderma fibrosis[J].Curr Opin Rheumatol,2009,21(6):623-629.
[13]Del GF,Sotgia F,de Almeida CJ,et al.Decreased expression of caveolin 1 in patients with systemic sclerosis:crucial role in the pathogenesis of tissue fibrosis[J].Arthritis Rheum,2008,58(9):2854-2865.
[14]Ihn H.Autocrine TGF-beta signaling in the pathogenesis of systemic sclerosisJ.J Dermatol Sci2008492103-113.
[15]Bonniaud P,Margetts PJ,Ask K,et al.TGF-beta and Smad3 signaling link inflammation to chronic fibrogenesis[J].J Immunol,2005,175(8):5390-5395.
[16]Luzina I G,Kopach P,Lockatell V,et al.Interleukin-33 potentiates bleomycin-induced lung injury[J].Am J Respir Cell Mol Biol,2013,49(6):999-1008.
[17]Yamamoto T,Takagawa S,Katayama I,et al.Effect of superoxide dismutase on bleomycin-induced dermal sclerosis:implications for the treatment of systemic sclerosis[J].J Invest Dermatol,1999,113(5):843-847.
[18]Nuovo GJ,Hagood JS,Magro CM,et al.The distribution of immunomodulatory cells in the lungs of patients with idiopathic pulmonary fibrosis[J].Mod Pathol,2012,25(3):416-433.
[19]Yamashita M,Iwama N,Date F,et al.Macrophages participate in lymphangiogenesis in idiopathic diffuse alveolar damage through CCL19-CCR7 signal[J].Hum Pathol,2009,40(11):1553-1563.
[20]Christmann RB,Sampaio-Barros P,Stifano G,et al.Association of Interferon-and transforming growth factor beta-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis[J].Arthritis Rheumatol,2014,66(3):714-725.
[21]Bhattacharyya S,Kelley K,Melichian DS,et al.Toll-like receptor 4 signaling augments transforming growth factorbeta responses:a novel mechanism for maintaining and amplifying fibrosis in scleroderma[J].Am J Pathol,2013,182(1):192-205.
[22]Park EJ,Roh J,Kim SN,et al.A single intratracheal instillation of single-walled carbon nanotubes induced early lung fibrosis and subchronic tissue damage in mice[J].Arch Toxicol,2011,85(9):1121-1131.
[23]Sueblinvong V, Neujahr DC, Mills ST,et al.Predisposition for disrepair in the aged lung[J].Am J Med Sci,2012,344(1):41-51.
[24]Papakonstantinou E,Aletras AJ,Roth M,et al.Hypoxia modulates the effects of transforming growth factor-beta isoforms on matrix-formation by primary human lung fibroblasts[J].Cytokine,2003,24(1-2):25-35.
[25]Corbel M,Caulet-Maugendre S,Germain N,et al.Inhibition of bleomycin-induced pulmonary fibrosis in mice by the matrix metalloproteinase inhibitor batimastat[J].J Pathol,2001,193(4):538-545.
[26]Grigoryev DN,Mathai SC,Fisher MR,et al.Identification of candidate genes in scleroderma-related pulmonary arterial hypertension[J].Transl Res,2008,151(4):197-207.
[27]Danczak-Pazdrowska A,Kowalczyk M,Szramka -Pawlak B,et al.Interleukin-17A and interleukin-23 in morphea[J].Arch Med Sci,2012,8(6):1089-1095.
猜你喜欢
中老年保健(2022年2期)2022-11-25
昆明医科大学学报(2022年4期)2022-05-23
昆明医科大学学报(2021年8期)2021-08-13
云南医药(2021年3期)2021-07-21
国际呼吸杂志(2019年21期)2019-11-25
电子制作(2019年12期)2019-07-16
电子制作(2018年8期)2018-06-26
电子测试(2017年11期)2017-12-15
中国现代医学杂志(2015年26期)2015-12-23
天津医科大学学报(2015年3期)2015-06-05
