
组织激肽释放酶对糖尿病大鼠局灶性脑缺血再灌注后炎症损伤的干预作用
2015-05-07 02:58石瑞峰曹琴琴赵玲玲张仁良
医学研究生学报 2015年9期
石瑞峰,刘 玲,胡 斌,陈 昕,曹琴琴,赵玲玲,张仁良
0 引 言
脑卒中是世界范围内致死率和致残率极高的疾病之一[1]。糖尿病已成为公认的缺血性脑卒中的独立危险因素。据估计,糖尿病患者发生脑卒中的风险较非糖尿病患者高1.5 ~3 倍。有研究报道,8%~63%的非糖尿病患者和39%~83%的糖尿病患者发生脑缺血性事件的同时伴发高血糖[2-3]。临床研究证实,糖尿病患者脑卒中后的短期及长期预后均较非糖尿病患者差,并且病死率相对较高[4-5]。研究表明,炎症反应是加重糖尿病患者脑缺血再灌注损伤的重要机制之一[6]。因此,抑制脑缺血再灌注损伤过程中的炎症反应对糖尿病患者具有神经保护作用。大量动物实验研究表明,组织激肽释放酶通过抑制炎症、减少凋亡、促进神经及血管再生对脑缺血再灌注损伤发挥保护作用[7-9]。然而,大量研究发现,并发糖尿病大鼠脑组织缺血再灌注后对部分神经保护的药物或者缺血前后预处理有抵抗性[10-11];且组织激肽释放酶对于糖尿病并发脑卒中是否具有神经保护作用尚未有研究证实。因此,本实验旨在通过观察组织激肽释放酶对糖尿病大鼠脑缺血再灌注后的行为学、脑梗死面积百分比、脑水肿程度改变、炎症细胞激活和细胞黏附分子表达的影响,探讨组织激肽释放酶的神经保护作用,为组织激肽释放酶治疗并发脑卒中的糖尿病患者提供理论依据。
1 材料与方法
1.1 实验动物及分组 健康清洁级成年雄性SD大鼠(100 ~150 g),由我院比较医学科提供,实验动物合格证号:CXK(军)2007-012。将动物置于实验室适应性饲养1 周,自由进食、饮水,室温(23±2)℃,相对湿度55%,每天光照12 h。采用随机数字表法将大鼠分为假手术组、等渗盐水组和组织激肽释放酶组,每组16 只。缺血再灌注后,等渗盐水组及组织激肽释放酶组立即分别尾静脉注射等渗盐水0.5 mL/kg和组织激肽释放酶1.6×10-2PNAU/kg(广东天普生化医药股份有限公司),假手术组于术后尾静脉注射等渗盐水0.5 mL/kg。实验过程严格遵循科技部《关于善待实验动物的指导性意见》。
1.2 动物模型制作及组织标本制备
1.2.1 糖尿病模型制备 参照文献[12]制备糖尿病大鼠模型。各组大鼠适应性喂养1 周后过渡为高脂饲料,4 周后禁食12 h,后腹腔注射STZ(美国Sigma 公司)35 mg/kg,常规喂养14 d,分别于注射STZ后第3、7 及14 天用血糖仪测定尾尖血糖,测定前禁食8 h,3 次血糖均>16.7 mmol/L 为模型制备成功,未达标准或未到时间点死亡者,剔除后补充。
1.2.2 局灶性脑缺血再灌注模型制备 参照文献[13]采用longa 法进行。腹腔注射10%水合氯醛(3 mL/kg)麻醉,颈前正中切口2 cm,充分暴露并分离侧颈总、颈内及颈外动脉,用动脉夹分别夹闭颈总及颈内动脉,永久性结扎颈外动脉远心端,另一粗线暂时结扎颈外动脉近心端,在两结扎点之间剪一小口,将特制尼龙栓线插入后用丝线固定并解开近心端粗线,离断颈外动脉剪口,继续插入至颈总动脉分叉处时,松开颈内动脉夹,将栓线拉向下方使之与颈内动脉成一直线并顺颈内动脉方向插入,直至大脑中动脉与大脑前动脉分叉处(此时阻力增大,线头距颈总动脉分叉处约18 mm),松开颈总动脉夹,缺血90 min 后将栓线拔出至颈外动脉残端,实现再灌注。假手术组仅暴露颈总动脉而不插入栓线。造模过程中使用烤灯维持大鼠肛温37 ℃。
1.2.3 标本留取 每组3 只大鼠脑缺血90 min 再灌注24 h,经10%水合氯醛腹腔麻醉后开胸,从心尖处向主动脉弓注入0.9%氯化钠溶液250 mL 行心脏灌注,随后用4%多聚甲醛250 mL(溶解于0.1 mol/L PBS 中,pH=7.4)灌注,固定后取脑,置于4%多聚甲醛后固定8 h,随后于30%蔗糖溶液中脱水沉糖24 h,经OCT 包埋剂包埋后行冰冻切片。
1.3 观察指标
1.3.1 神经功能缺损评分 每组分别随机取6 只大鼠,参照改良神经功能缺损评分(modified Neurological Severity Scores,NSS)[14]行神经功能评价:0分为正常;1 ~6 分为轻度神经功能损伤;7 ~12 分为中度神经功能损伤;13 ~18 分为重度神经功能损伤。在模型制作后24 h 进行评分。0 分者及未到24 h 死亡者剔除,随机补充。
1.3.2 脑梗死面积测定 每组分别随机取4 只大鼠,缺血再灌注24 h 后断头取脑,沿冠状位从额极至枕叶以厚度2 mm/片切成6 片,TTC 磷酸盐缓冲液中,37 ℃避光孵育15 min,随后用4%多聚甲醛固定。扫描并用Image-J 测定每张脑片梗死面积,并计算梗死面积百分比。
1.3.3 脑水肿测定 每组分别随机取6 只大鼠,缺血再灌注24 h 后断头取脑,取梗死侧半球于精密电子天平称重为湿重(WW),随后将脑组织置入100 ℃烘箱中烘烤24 h,再次称重记录为干重(DW),计算公式如下:
脑水肿程度=[(WW-DW)/WW]×100%
1.3.4 小胶质细胞离子钙接头蛋白(ionized calcium bindingadaptor molecule-1,Iba1)及髓过氧化物酶(myeloperoxidase,MPO)免疫组化染色 切片置于4%多聚甲醛中固定15 min 后,于0.3%H2O2浸泡30 min,随后置于0.1%Triton 15 min,加5%牛血清蛋白封闭1 h;分别滴加兔抗鼠Iba1 抗体(1∶1000)及MPO抗体(1∶200),4 ℃,24 h;滴加生物素化羊抗兔IgG二抗(1∶200),37 ℃,2 h;滴加SABC 后用DAB-H2O2显色。每步之间用0.1 mol/L PBS(pH=7.4)冲洗3次,每次5 min。常规脱水,透明,封固。
1.3.5 脑组织血管细胞黏附分子-1(vascular cell adhesion molecule 1,VCAM-1)及缺血半暗带区细胞间黏附分子-1(intercellular adhesion molecule 1,ICAM-1)mRNA 表达测定 采用RT-PCR 方法测定脑组织中VCAM-1 及ICAM-1 表达水平。每组分别取3 只大鼠,再灌注24 h 后断头取脑,取缺血半暗带皮质区标本采用Trizol 抽提RNA,通过逆转录将总RNA 反转为cDNA,使用PCR 仪扩增,扩增使用引物序列如下:VCAM-1:5'-TGCTCCTGACTTGCAGCACCAC-3',5'-TGTCATCGTCACAGCAGCACCC-3';ICAM-1:5'-TGCAGCCGGAAAGCAGATGGTG-3',5'-ATGGACGCCACGATCACGAAGC-3'; GAPDH: 5'-GCAAGTTCAACGGCACAG-3',5'-GCCAGTAGACTCCACGACAT-3'。
1.4 统计学分析 应用SPSS 21.0 软件进行统计分析。定量资料以均数±标准差()表示,行为学采用Kruskal-Wallis 非参数检验,多重比较经Bonferroni 校正;多个均数间比较采用单因素方差分析,两两比较采用LSD 检验;方差不齐时行Welch 校正的Dunnett'sT3 检验。以P≤0.05 为差异有统计学意义。
2 结 果
2.1 神经功能缺损评分 假手术组大鼠无明显神经功能缺损,等渗盐水组及组织激肽释放酶组大鼠均有不同程度的神经功能缺损,且组织激肽释放酶组大鼠的神经功能缺损评分明显低于等渗盐水组(P <0.01)。见图1。
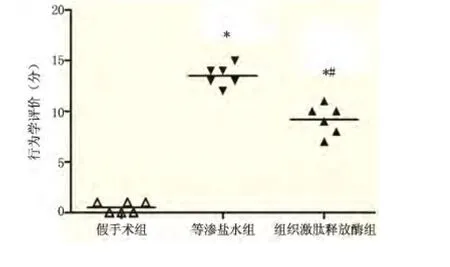
图1 大鼠脑缺血再灌注损伤后神经功能缺损评分比较Figure 1 Neurological Severity Scores of different groups of rats 24 h after MCAO
2.2 大鼠脑梗死面积百分比 等渗盐水组及组织激肽释放酶组经TTC 染色梗死区呈苍白色,主要位于额顶叶皮质及纹状体(即MCA 供血区),未缺血区呈红色。假手术组未见梗死区域,组织激肽释放酶组梗死面积百分比较等渗盐水组显著降低(P <0.05)。见表1,图2。
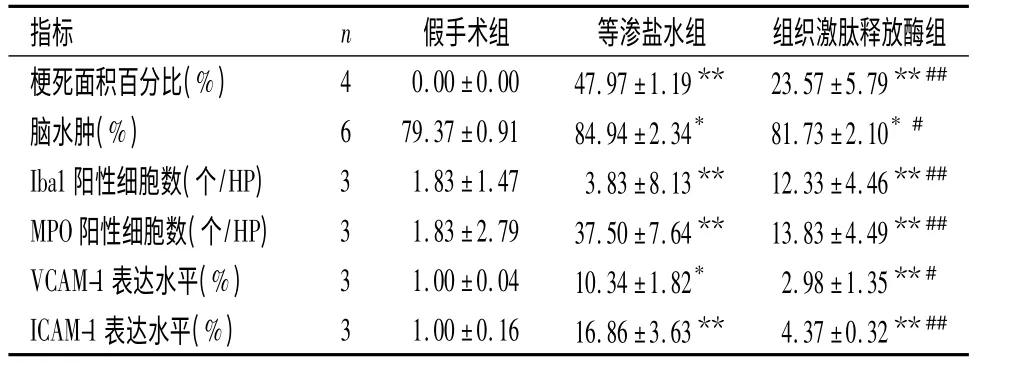
表1 大鼠各项观察指标比较Table 1 Infarct area,brain edema,Iba1-and MPO-positive cells,and expressions of VCAM-1 and ICAM-1 in different groups of rats
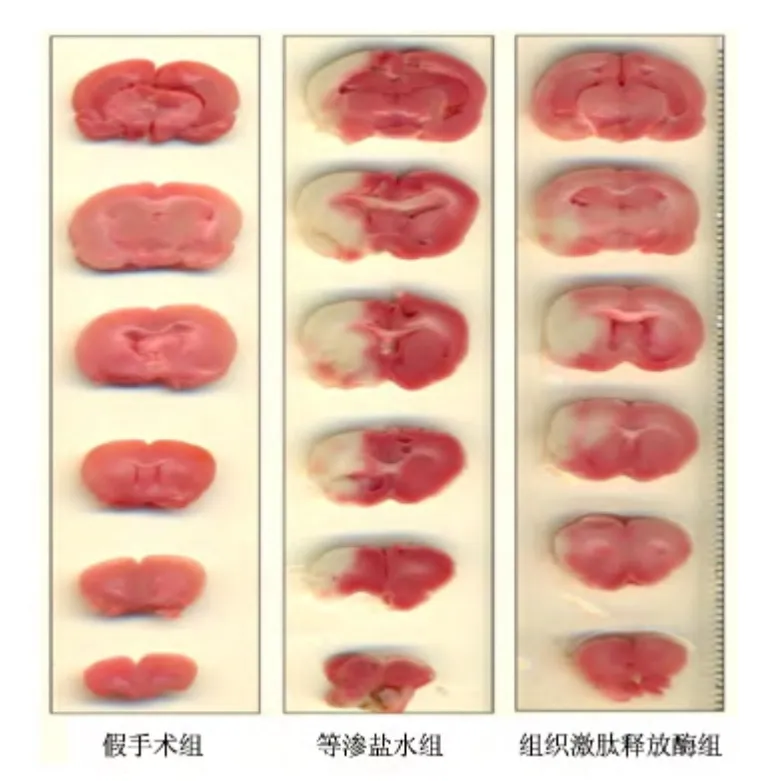
图2 TTC 染色观察各组大鼠脑梗死面积Figure 2 TTC staining for different groups of rats at 24 h after MCAO
2.3 大鼠脑水肿程度比较 脑水肿是反映脑组织早期炎症反应的重要指标之一。等渗盐水组脑水肿程度高于假手术组(P <0.05),并且组织激肽释放酶组脑水肿程度低于等渗盐水组(P <0.05)。见表1。
2.4 小胶质细胞及髓过氧化物酶免疫组化染色Iba1 是小胶质细胞的标志性蛋白,卒中后炎症反应导致其呈活化状态。假手术组双侧大脑半球、等渗盐水组和组织激肽释放酶组缺血对侧半球少见Iba1 阳性细胞,缺血半暗带区Iba1 阳性细胞表达较多,缺血核心区Iba1 阳性细胞表达较少。等渗盐水组Iba1 阳性细胞表达显著高于假手术组(P <0.05),并且组织激肽释放酶治疗组Iba1阳性细胞表达较等渗盐水对照组显著降低(P <0.05)。见表1,图3。
MPO 是中性粒细胞活化标志蛋白,缺血再灌注后梗死边缘区MPO 表达显著增多,假手术组未见阳性细胞,等渗盐水组见大量MPO 免疫染色阳性的中性粒细胞,经组织激肽释放酶治疗后MPO 表达显著降低(P <0.05)。见表1,图4。

图4 高倍镜下观察各组大鼠脑组织中MPO 阳性细胞(免疫组化染色 ×200)Figure 4 Count of MPO-positive cells wascounted in different groups of rats at 24 h after MCAO(IHC ×200)
2.5 VCAM-1 及ICAM-1mRNA 表达 缺血再灌注24 h 后,各组缺血半暗带皮质区VCAM-1 及ICAM-1 表达水平存在显著性差异。与假手术组相比,等渗盐水对照组VCAM-1、ICAM-1 表达水平提高(P <0.05);与等渗盐水对照组相比,组织激肽释放酶治疗组VCAM-1 及ICAM-1 表达水平显著降低(P <0.05)。见表1。
3 讨 论
高血糖不仅是脑卒中发生的独立危险因素,也是加重脑卒中不良预后的主要原因。单纯控制糖尿病患者血糖并不能提高卒中后神经功能的恢复,急性期内血管再通仍为首选治疗方法。然而,急性期血管内再通导致缺血再灌注损伤,加重局部炎症反应及神经细胞损伤。
在脑缺血病理过程中,谷氨酸介导的兴奋性中毒、钙离子超载、氧化应激反应、压力信号通路、神经血管病理生理反应及炎症、细胞凋亡及坏死等多种因素均发挥重要作用[15]。机体在高血糖状态下,不仅出现细胞及内部功能紊乱导致代谢异常,基因的表达也会出现异常变化,加重缺血后脑组织损伤[16]。研究表明,高血糖通过激活核因子-κB 提高黏附分子的表达,进而促进白细胞的黏附[3]。另外,高血糖能加速高迁移率蛋白-1 的释放从而促进缺血后脑组织中星形胶质细胞及小胶质细胞的活化,加剧炎症反应[17]。
组织激肽释放酶是激肽释放酶-激肽系统(kallikrein-kinin system,KKS)的成员之一。作为体内重要的炎性调节系统,KKS 主要包括激肽释放酶、激肽原和激肽,激肽原在激肽释放酶的作用下转化为激肽和缓激肽,后者作用于缓激肽受体1(bradykinin 1 receptor,B1R)和/或受体2(B2R)或蛋白酶活化受体(protease activated receptors,PARs),激肽释放酶广泛存在于组织及血浆之中,存在于组织中者称为组织激肽释放酶。大量研究已证实,组织激肽释放酶可对多种炎症细胞发挥抑制作用,抑制缺血再灌注损伤激活的细胞因子和黏附分子表达,从而抑制炎症反应[7]。缓激肽受体在糖尿病大鼠和非糖尿病大鼠脑缺血再灌注后表达及作用由明显差异。在本研究中,与等渗盐水组相比,组织激肽释放酶组大鼠的神经功能缺损评分显著改善、梗死面积减小、脑水肿程度明显降低,表明组织激肽释放酶对糖尿病脑缺血再灌注起显著的神经保护作用。本实验采用2 型糖尿病大鼠模型制备方法,经高脂饲料喂养与低剂量STZ 联合诱导,以模仿临床常见的2 型糖尿病病理模型[10]。缺血再灌注模型采用经典longa 线栓法诱导,以模仿临床急性脑卒中患者溶栓或取栓后脑组织缺血再灌注损伤病理生理过程[11]。Chen 等[8]研究通过对大鼠脑缺血再灌注给予组织激肽释放酶后不同时间点的研究已表明,24 h 后大鼠行为学、脑水肿、凋亡及炎症水平均出现显著统计学差异,因为本实验研究选取给药后24 h 作为标本采集时间点。
小胶质细胞作为中枢神经系统的“清道夫”,在中枢神经系统损伤时起重要作用,然而过度激活的小胶质细胞会分泌大量细胞因子和细胞毒性物质,引发神经毒性[18]。在本实验研究发现,经组织激肽释放酶治疗后的大鼠脑组织缺血半暗带区域激活的小胶质细胞显著低于等渗盐水组,这与Noda 等[19]的研究结果一致。该研究表明,缓激肽通过抑制小胶质细胞释放IL-1β 和TNF-α 调节炎症反应,从而发挥神经保护作用。
缺血后再灌注时,大量活化的白细胞在受损的内皮细胞表明滚动、黏附,最终渗入周围组织,释放蛋白酶、氧自由基等物质加重内皮损伤,改变内皮通透性,加重血脑屏障的破坏,进而加重脑组织水肿且减少再灌注血流。渗入组织的白细胞通过呼吸爆发及溶酶体酶的释放进一步加重缺血脑组织损伤[20]。MPO 作为中性粒细胞的标志性蛋白,可评估活化中性粒细胞在组织中的迁移[21]。本研究发现,缺血再灌注损伤后,MPO 阳性染色细胞数目增多,经组织激肽释放酶治疗后明显减少,表明早期组织激肽释放酶治疗可减轻炎症介导的缺血性神经功能损害。
黏附分子作为卒中后抗炎药物靶点,在脑卒中后的白细胞迁移过程中发挥重要作用。上已述及,白细胞从血管渗入脑组织需经过滚动、黏附以及游出3 个步骤。细胞黏附分子作为介导白细胞聚集、游出血管的关键性物质主要包括3 种:选择素、免疫球蛋白超家族和整合素。免疫球蛋白超家族包括5种分子,其中ICAM-1 在内皮细胞、白细胞膜表面持续低水平表达,细胞因子刺激能使其表达上调,VCAM-1 在肿瘤坏死因子-α 和白细胞介素-1 的诱导下表达上调[22-23]。本实验研究发现,糖尿病大鼠缺血再灌注后,脑组织内ICAM-1 及VCAM-1 mRNA表达均显著升高,而再灌注早期给予组织激肽释放酶治疗可明显降低上述黏附分子的表达水平,从而抑制白细胞的游走渗出,减轻炎症反应。
综上所述,本实验显示组织激肽释放酶对STZ诱导的糖尿病大鼠脑缺血再灌注损伤模型具有神经保护作用。组织激肽释放酶通过降低ICAM-1 和VCAM-1 mRNA 的表达减少中性粒细胞的浸润,同时抑制小胶质细胞的激活,从而减轻缺血再灌注引发的炎症反应,减少梗死面积,减轻脑水肿,从而改善神经功能。因此,组织激肽释放酶发挥神经保护作用机制可能与抑制脑缺血再灌注局部炎症反应有关。本实验证实组织激肽释放酶对抑制糖尿病大鼠脑缺血再灌注局部炎症有效,有望为糖尿病患者卒中早期临床用药提供实验依据。
[1] 晏正辉,王兴元.老年人急性脑血管疾病的脑电图特征[J].医学研究生学报,2013,26(6):615-617.
[2] 宋佳希,汪俊军.军队人员心血管疾病危险评估的研究进展[J].医学研究生学报,2013,26(10):1088-1091.
[3] Kruyt ND,Biessels GJ,Devries JH,et al.Hyperglycemia in acute ischemic stroke:pathophysiology and clinical management[J].Nat Rev Neurol,2010,6(3):145-155.
[4] Kimura K,Sakamoto Y,Iguchi Y,et al.Admission hyperglycemia and serial infarct volume after t-PA therapy in patients with and without early recanalization.[J].J Neurol Sci,2011,307(1-2):55-59.
[5] Luitse MJ,Biessels GJ,Rutten GE,et al.Diabetes,hyperglycaemia,and acute ischaemic stroke[J].Lancet Neurol,2012,11(3):261-271.
[6] Panés J,Kurose I,Rodriguez-Vaca D,et al.Diabetes exacerbates inflammatory responses to ischemia-reperfusion[J].Circulation,1996,93(1):161-167.
[7] Albert-Weiβenberger C,Sirén AL,Kleinschnitz C.Ischemic stroke and traumatic brain injury:The role of the kallikrein-kininsystem[J].Prog Neurobiol,2013,101(4):65-82.
[8] Chen ZB,Huang DQ,Niu FN,et al.Human urinary kallidinogenase suppresses cerebral inflammation in experimental stroke and downregulates nuclear factor-κB[J].J Cereb Blood Flow Metab,2010,30(7):1356-1365.
[9] Ling L,Hou Q,Xing S,et al.Exogenous kallikrein enhances neurogenesis and angiogenesis in the subventricular zone and the peri-infarction region and improves neurological function after focal cortical infarction in hypertensive rats[J].Brain Res,2008,1206(3):89-97.
[10] Li D,Huang B,Liu J,et al.Decreased brain K(ATP)channel contributes to exacerbating ischemic brain injury and the failure of neuroprotection by sevoflurane post-conditioning in diabetic rats[J].PLoS One,2013,8(8):e73334.
[11] Kersten JR,Toller WG,Gross ER,et al.Diabetes abolishes ischemic preconditioning:role of glucose,insulin,and osmolality[J].Am J Physiol-Heart C,2000,278(4):H1218-H1224.
[12] Chen GM,Hu N,Liu L,et al.Pharmacokinetics of verapamil in diabetic rats induced by combination of high-fat diet and streptozotocin injection[J].Xenobiotica,2011,41(6):494-500.
[13] Su J,Cui M,Tang Y,et al.Blockade of bradykinin B2 receptor more effectively reduces postischemic blood-brain barrier disruption and cytokines release than B1 receptor inhibition[J].Biochem Bioph Res Co,2009,388(2):205-211.
[14] Chen J,Li Y,Wang L,et al.Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats[J].Stroke,2001,32(4):1005-1011.
[15] Mehta SL,Manhas N,Raghubir R.Molecular targets in cerebral ischemia for developing novel therapeutics[J].Brain Res Rev,2007,54(1):34-66.
[16] Muranyi M,Fujioka M,He Q,et al.Diabetes activates cell death pathway after transient focal cerebral ischemia.[J].Diabetes,2003,52(2):481-486.
[17] Huang J,Liu B,Yang C,et al.Acute hyperglycemia worsens ischemic stroke-induced brain damage via high mobility group box-1 in rats[J].Brain Res,2013,1535(42):148-155.
[18] Baron JC,Yamauchi H,Fujioka M,et al.Selective neuronal loss in ischemic stroke and cerebrovascular disease.[J].J Cerebr Blood F Met,2014,34(1):2-18.
[19] Noda M,Kariura Y,Pannasch U,et al.Neuroprotective role of bradykinin because of the attenuation of pro-inflammatory cytokine release from activated microglia.[J].J Neurochem,2007,101(2):397-410.
[20] Perez-De-Puig I,Miró-Mur F,Ferrer-Ferrer M,et al.Neutrophil recruitment to the brain in mouse and human ischemic stroke[J].Acta Neuropathol,2015,129(2):239-257.
[21] Ling LK,Zhi YW,Ning H,et al.Neutralization of chemokinelike factor 1,a novel CC chemokine,protects against focal cerebral ischemia by inhibiting neutrophil infiltration via MAPK pathways in rats[J].J Neuroinflamm,2014,11(1):1-10.
[22] Wang Q,Tang XN,Ma Y.The inflammatory response in stroke.[J].J Neuroimmunol,2007,184(1-2):53-68.
[23] Lin M,Sun W,Gong W,et al,Methylophiopogonanone a protects against cerebral ischemia/reperfusion injury and attenuates blood-brain barrier disruption in vitro[J].PLoS One,2015,10(4):e0124558.
猜你喜欢
昆明医科大学学报(2020年11期)2020-12-28
中学生数理化·八年级物理人教版(2019年12期)2019-05-21
中成药(2018年4期)2018-04-26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中成药(2017年6期)2017-06-13
小猕猴智力画刊(2016年8期)2016-05-14
中国康复理论与实践(2015年10期)2015-12-24
少儿科学周刊·儿童版(2015年11期)2015-12-17
中国体外循环杂志(2015年3期)2015-12-08
医学研究杂志(2015年12期)2015-06-10
