
假肥大型肌营养不良两家系产前诊断及遗传咨询
2015-05-05 09:50熊盈钟燕芳潘小英刘玲
中国产前诊断杂志(电子版) 2015年4期
熊盈 钟燕芳 潘小英 刘玲
(广东省妇幼保健院,广东 广州 510010)
假肥大型肌营养不良两家系产前诊断及遗传咨询
熊盈 钟燕芳*潘小英 刘玲
(广东省妇幼保健院,广东 广州 510010)
目的 通过描述两例假肥大型肌营养不良家系病例,探讨该病的遗传咨询及产前诊断。方法 对两例假肥大型肌营养不良的先证者利用多探针连接依赖的扩增技术(multiplex ligation-dependent probe amplification, MLPA)方法基因确诊,遗传咨询后,分别于孕13+周和孕21+周抽取绒毛和羊水行胎儿产前诊断,明确胎儿基因情况。结果 两例胎儿产前诊断的结果分别为未检出母亲缺失基因突变和DMD基因45-53纯合突变,其中后一例选择终止妊娠。结论 通过及时的产前诊断及有效的遗传咨询,可减少假肥大型肌营养不良患者的出生,达到优生优育的目的。
假肥大型肌营养不良;产前诊断;遗传咨询
假肥大型肌营养不良包括Duchenne型肌营养不良(Duchenne muscular dystrophy,DMD)和Becker型肌营养不良(Becker muscular dystrophy,BMD)。DMD是最常见的X连锁隐性遗传的疾病,发病率约1/3500活产男婴,而BMD的发病率约为DMD患者的1/10。 DMD/BMD是编码抗肌萎缩蛋白(Dystrophin)的基因(DMD/BMD)异常所引起的一类疾病,主要的临床表现为进行性肌肉无力及相关并发症。目前针对DMD/BMD的治疗方法仍很局限,预防该病患者的出生是控制此类出生缺陷的主要措施。本文报道通过两假肥大型肌营养不良家系,对该病的产前诊断及遗传咨询进行探讨。
1 对象和方法
1.1 基本资料
1.1.1 病例1 女,27岁,G2P1,孕12+周,因曾生育DMD患儿就诊。该孕妇3年前足月剖宫产一子,其半岁时因持续发热住院治疗,入院后查肌酶异常:肌酸激酶(CK)6 389 U/L,心肌型肌酸激酶同工酶(CK-MB)186 U/L,外周血检查提示为DMD45-52纯合子性突变。目前生长发育尚可,可自己行走。家族史:父母适龄结婚,无神经肌肉疾病病史,其弟7岁开始出现进行性走路不稳,腓肠肌肥大,呈典型“鸭步”,逐渐不能行走,20岁时因循环衰竭、呼吸道感染死亡,未行基因检查。抽取孕妇外周血DMD基因检查,结果提示为DMD45~52杂合缺失突变,为DMD基因携带者。遗传咨询后,孕妇于孕13+周行绒毛取样术查胎儿DMD基因情况。
1.1.2 病例2 女,42岁,G4P1,孕20+周,因高龄妊娠、唐筛高风险就诊。询问家族史发现其弟15岁时出现双下肢肌力下降,走路伴有轻微晃动,下肢腓肠肌肥大,无上肢及其余部位肌力异常表现,目前38岁,能独立行走,日常生活未受明显影响。孕妇及其弟抽取外周血行DMD基因检查,结果提示本人为第45~53外显子杂合缺失,为DMD携带者;而其弟为DMD基因第45~53外显子纯合缺失。遗传咨询后,孕妇于孕21+周行羊膜腔穿刺术抽取羊水送胎儿DMD基因及胎儿染色体检查。
1.2 方法
1.2.1 取得标本及DNA提取 两孕妇分别于孕13+周和孕21+周在本中心由经验丰富医生在超声引导下抽取胎儿绒毛(8支)和羊水(10 ml)。使用Qiagen公司试剂盒提取基因组DNA,提取方法参照说明书,并置于-20℃保存。
1.2.2 串联重复序列(short tandem repeat,STR) 对D13S305、D18S978、D21S11等11个STR位点测序分析有无母体DNA污染。
1.2.3 MLPA方法 MLPA试剂盒SALSA P034、P035(MRC-Holland公司)检测目标基因的79个外显子突变情况,每批实验均设立正常男、女性对照,分析软件得出结论,具体操作方法参照说明书。正常峰值比(0.7≤峰值比≤1.3),峰值比>1.3提示基因重复,峰值比<0.7提示基因缺失。
2 结 果
病例一中先证者母亲这一胎绒毛检查结果提示胎儿在检测范围内未遗传到母亲的基因缺失突变(图1),STR未检出异常。目前孕妇定期产检,无特殊情况。
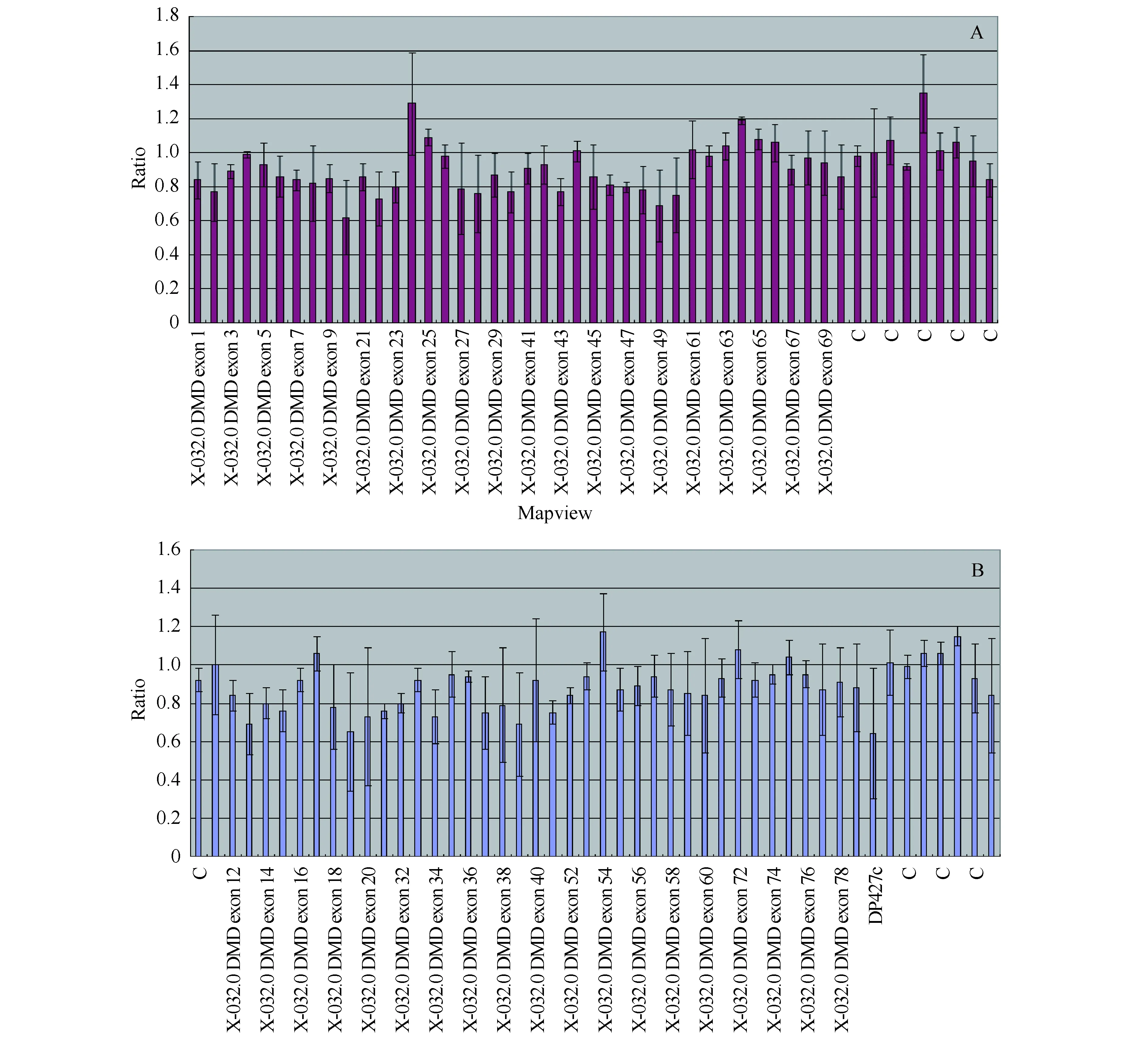
图1 A和B分别病例一MLPA P034、P035探针检测结果
病例二中孕妇本胎羊水检查结果提示胎儿为DMD基因45~53纯合突变(图2),胎儿染色体核型未见明显异常。遗传咨询后,孕妇夫妇决定终止妊娠。
3 讨 论
DMD/BMD基因位于染色体Xp21,跨度2300kb,含79个外显子。其主要的异常包括缺失、 重复和点突变,其中约65%为基因缺失,该基因中段的44~53外显子和5’端的第2~20外显子是发生缺失和重复的热点区域。临床表型与基因缺失的大小并没有明显相关性,而多数取决于基因缺失是否破坏了阅读框[1,2]。抗肌萎缩蛋白是细胞骨架的组成成分,在骨骼肌和心肌等细胞膜表面发挥作用,是肌纤维维持稳定的重要元素。当基因缺失造成移码突变,不能编码功能正常的抗肌萎缩蛋白时,临床表型为DMD:3~5岁起病,骨盆带肌肉无力,呈典型“鸭步”,跟腱挛缩、双足下垂,多于12岁左右丧失行走能力;肩胛肌受累,翼状肩胛,举臂无力;肌肉假性肥大,以腓肠肌最为明显;部分患儿出现不同程度智力障碍;心肌损害,多数病人于20~30岁死于呼吸及循环系统衰竭[3]。当基因缺失并未造成阅读框破坏,仍能编码有功能的抗肌萎缩蛋白时,临床表型为BMD:被认为是DMD的一种不典型类型,症状与之相似,但发病较晚,症状较轻,心脏少受累,少数可接近正常生命年限。两者致病基因相同,临床特征主要区别于DMD依赖轮椅的时间多在13岁前,而BMD却常在16岁以后,甚至部分患者在40岁以后行走功能仍只轻微受影响。

图2 A和B分别病例二MLPA P034、P035探针检测结果
在本文的第一个病例中,先证者为DMD45~52纯合子性突变,其舅舅为典型DMD临床表现, 这与之前的文献报道相符[2]。在第二个病例中,45~53外显子纯合缺失先证者的临床症状较轻,发病时间较晚,目前已38岁仍能正常生活,符合BMD临床诊断。以往的文献中,外显子45~53缺失异常的临床表现较复杂,有DMD和BMD两种表型,并不完全符合阅读框假说,可能与缺失范围大、突变的氨基酸不能维持功能、外显子自发跳跃等原因有关[2,4]。
DMD是最常见的X连锁隐性遗传的肌病,女性携带者与正常男性生育男性患儿的风险为50%,而生育女性后代则有50%携带致病基因的风险;男性患者与正常女性生育男性后代患病的风险为0%,而生育女性后代则100%为携带者。约2/3的患者来源于母亲遗传,呈明显家族性发病,另有1/3为散发病例,多为新发基因突变所致。对于携带此基因的女性,大多数不发病,然而有部分携带者会出现轻重不一的临床表现,这可能与正常基因所在的染色体在胚胎早期灭活导致细胞功能异常等原因有关。
由于目前对DMD患者尚无有效治疗方法,控制患病胎儿的出生显得尤为重要。先证者经基因确诊后,对其进行遗传咨询,建议行母系基因检测,明确该患者是来源于母系遗传还是新发突变。若能明确母亲携带者,告知发病规律,在其下次妊娠时可行介入性产前诊断明确下一胎DMD基因情况。若为新发突变,告知再发风险由孕妇自行决定是否行产前诊断。目前最早可在妊娠11~13周间行胎儿绒毛取样术送胎儿DMD基因检查,或于18周后行羊膜腔穿刺术抽取羊水送检,必要时可于孕24周后抽取脐血。产前诊断过程中需高度警惕标本污染可能,STR位点测序不仅可以对判断标本是否存在母源污染及污染程度,还可同时检测21、18、13以及X/Y染色体的数目,对发现常见染色体数目异常有帮助。
在遗传咨询过程中详细了解患者家族史尤为重要。在第二个病例中,孕妇并非因DMD/BMD就诊,若病史采集过程中疏忽了家族史,羊膜腔穿刺只为胎儿染色体检查,本例肌营养不良胎儿则会被漏诊。准确诊断先证者、明确家系基因情况是进行遗传咨询和产前诊断的前提,是防控DMD/BMD的重要手段,有利于减轻了整个社会和家庭的沉重负担。
[1] Monaco AP, Bertelson CJ, Liechti-Gallati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus[J].Genomics,1988,2:90-95.[2] 冯善伟,梁颖茵,操基清,等.假肥大型肌营养不良症的基因型与临床表型关系分析[J],中华医学遗传学杂志,2012,29(6): 653-657.
[3] Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy[J]. Lancet Neurol, 2010,9(1):77-93.
[4] Bosone II, Bortolotto S, Mongini T, et al. Late onset and very mild course of Xp21 Becker type muscular dystrophy[J]. Clin Neuropathol,2001,20(5):196-199.
编辑:宋文颖
Objective To discuss the prenatal diagnosis and genetic counselling in two families with pseudohypertrophic muscular dystrophy. Method We detected gene mutation in two propositus and collected the chorionic villus at 13 weeks of gestation and amniotic fluid cell at 21 weeks of gestation to analyze whether there was gene mutation in fetus. Resuls The first fetus was not detected the gene mutation; however we found the homozygous gene mutation in second fetus which was induced labor. Conclusions We can prevent the birth of the fetus with pseudohypertrophic muscular dystrophy through effective prenatal diagnosis.
pseudohypertrophic muscular dystrophy; prenatal diagnosis; genetic consultation
10.13470/j.cnki.cjpd.2015.04.011
R714.55
A
2015-09-13)
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
现代临床医学(2022年4期)2022-09-29
临床输血与检验(2022年3期)2022-06-22
山东医药(2021年24期)2021-09-01
中老年保健(2021年12期)2021-08-24
中国生殖健康(2020年4期)2021-01-18
中西医结合肝病杂志(2020年2期)2020-10-27
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
中国生殖健康(2018年4期)2018-11-06
