
高效液相色谱-串联质谱法测定豆芽中8种药物残留
2015-04-27 03:15张亚莲王岁楼徐牛生余可垚沈伟健赵增运沈崇钰
分析测试学报 2015年2期
张亚莲,柳 菡,王岁楼*,丁 涛*,徐牛生,余可垚,沈伟健,赵增运,刘 芸,吴 斌,张 睿,沈崇钰
(1.中国药科大学 药学院,江苏 南京 210009;2.江苏出入境检验检疫局,江苏 南京 210001;3.赛默飞世尔科技色谱质谱部,上海 201206)
高效液相色谱-串联质谱法测定豆芽中8种药物残留
张亚莲1,柳 菡2,王岁楼1*,丁 涛2*,徐牛生3,余可垚2,沈伟健2,赵增运2,刘 芸2,吴 斌2,张 睿2,沈崇钰2
(1.中国药科大学 药学院,江苏 南京 210009;2.江苏出入境检验检疫局,江苏 南京 210001;3.赛默飞世尔科技色谱质谱部,上海 201206)
建立了豆芽中8种药物(4-氯苯氧乙酸、吲哚乙酸、吲哚丁酸、1-萘乙酸、6-苄基腺嘌呤、2,4-二氯苯氧乙酸、赤霉素和多菌灵)残留的高效液相色谱-串联质谱(LC-MS/MS)检测方法。豆芽样品经0.1%冰醋酸-乙腈溶液提取、浓缩,分散固相萃取剂净化后,用液相色谱-串联质谱测定,外标法定量。8种药物在5~100 μg/L范围内呈良好的线性关系(r2>0.99),定量下限为5 μg/kg。在5,10,20 μg/kg 3个加标水平下,8种药物的回收率为71.6%~87.9%,相对标准偏差不大于14.6%。方法准确、简单、快速,可用于豆芽中8种药物的同时测定。
高效液相色谱-串联质谱法(LC-MS/MS);豆芽;药物残留
豆芽是日常生活中极为常见的一种蔬菜,其生产过程简单,成本低、利润大。不少商贩纷纷转向豆芽生产,但市场上的豆芽货源混乱。一些商贩为获取高额利润,在豆芽生产过程中使用一些药物(如“无根粉”、“速长剂”、“AB粉”等),导致毒豆芽事件时有发生。在豆芽生长中常用的药物有4-氯苯氧乙酸、吲哚乙酸、吲哚丁酸、1-萘乙酸、6-苄基腺嘌呤、2,4-二氯苯氧乙酸、赤霉素和多菌灵等。这些药物大部分属于植物生长调节剂,是一类用于调节植物生长发育的农药,可促进植物生长、提高农作物的质量和产量。如4-氯苯氧乙酸(又名防落素)是一种有机酸,可防止植物落花、落果,将其以4-氯苯氧乙酸钠的形式用于豆芽生产中,可抑制豆芽生根并催熟生长;6-苄基腺嘌呤是细胞分裂素的一种,可促进细胞分裂,诱导组织分化,用于豆芽生产中可促进芽的生长,抑制根的生长,缩短生长周期[1];2,4-二氯苯氧乙酸主要用于诱导细胞增殖,引起脱分化和未组织化的细胞生长,对豆芽生长具有很好的促进和调节作用;赤霉素是广泛存在的一类植物激素,可刺激叶和芽的生长,在植物幼苗生长过程中用于提高发芽率,可提高豆芽产量。但近年来研究发现,植物生长调节剂虽然低毒,但会在农作物中残留并通过食物链进入人体,若长期食用会造成一定程度的伤害。GB2760-2011有关问题的复函[2]中明确指出,4-氯苯氧乙酸钠、6-苄基腺嘌呤等物质不得作为食品用加工助剂生产经营和使用。GB2760-2011[3]规定,2,4-二氯苯氧乙酸在经表面处理的新鲜蔬菜、鲜水果中的最大使用量为0.01 g/kg,最大残留量为2.0 mg/kg;其他植物生长调节剂未被收录其中。
目前,针对豆芽中4-氯苯氧乙酸、6-苄基腺嘌呤、2,4-二氯苯氧乙酸和赤霉素的检测方法主要有高效液相色谱法[4-10]和高效液相色谱-串联质谱法[1,11-14],气相色谱法[15]、气相色谱-串联质谱法[16-17]、离子色谱法[18]和薄层色谱法[19]也有报道;而豆芽中多菌灵的检测方法较少,有高效液相色谱法[9]和高效液相色谱-串联质谱法[11];已报道的豆芽中吲哚乙酸和吲哚丁酸的检测方法只有高效液相色谱法[20]。其中,高效液相色谱法和气相色谱法的灵敏度低、定性能力差、抗干扰能力弱;气相色谱-串联质谱法一般用于检测易挥发、弱极性的化合物,而上述几种药物均为难挥发、强极性的化合物,如采用此方法检测,需要进行衍生化;离子色谱法对非溶液样品的净化要求高;薄层色谱法的灵敏度差,定量误差大;高效液相色谱-串联质谱法的特异性强、灵敏度高、定性能力好,且不需对待测物进行衍生化。目前采用高效液相色谱-串联质谱法(LC-MS/MS)同时检测豆芽中8种药物(4-氯苯氧乙酸、吲哚乙酸、吲哚丁酸、1-萘乙酸、6-苄基腺嘌呤、2,4-二氯苯氧乙酸、赤霉素、多菌灵)的研究尚未见报道。
根据豆芽基质的特点,本文采用QuEChERS技术[21]从4个方面(提取方式、提取溶剂、盐的选择和用量、分散固相萃取剂的选择和用量)对上述8种药物回收率的影响进行考察,得到最优的前处理方法,并采用高效液相色谱-串联质谱法进行检测。方法简单、快速、灵敏,能够满足大批量豆芽样品中8种药物同时检测的要求。
1 实验部分
1.1 仪器与试剂
Accela高效液相色谱仪(Thermo公司)和TSQ Quantum Access三重四极杆串联质谱(Thermo-Fisher公司),配有Xcalibur 1.4版软件,KQ-250DE超声仪(昆山市超声仪器有限公司),XW-80A涡旋混匀器(上海医大仪器厂),LDZ5-2离心机(Sigma公司),Milli-Q去离子发生器(Millipore公司),真空氮气吹干仪(Caliper公司)。
4-氯苯氧乙酸(4-Chlorophenoxyacetic acid,4-CPA)、吲哚乙酸(Indole-3-acetic acid,IAA)、吲哚丁酸(Indole-3-butyric acid,IBA)、1-萘乙酸(1-Naphthlcetic acid,NAA)、6-苄基腺嘌呤(6-Benzylaminopurine,6-BAP)、2,4-二氯苯氧乙酸(2,4-Dichlorophenoxyacetic acid,2,4-D)、赤霉素(Gibberellin acid,GA3)、多菌灵(Carbendazim)8种药物标准物质纯度均大于99%,购于Sigma公司;甲醇、醋酸铵(色谱级,德国Merck公司);乙腈(分析级)、冰醋酸(南京化学试剂有限公司)。实验用水为去离子水。
实验用黄豆芽、绿豆芽样品均购自超市。
1.2 标准溶液的配制
标准储备液:分别准确称取适量的上述8种药物标准物质,用甲醇溶解并稀释配制成0.2 g/L的标准储备液,置于冰箱中0 ℃保存备用。分别准确量取适量的上述8种标准储备液,混合,用甲醇稀释配制成1 mg/L和0.1 mg/L的混合标准溶液,用于标准工作溶液的配制,并置于冰箱中0 ℃保存备用。
标准工作溶液:准确吸取一定量的混合标准溶液,用甲醇-水(体积比3∶7)混合溶液稀释至5,10,20,50,100 μg/L,作为标准工作液。
1.3 样品前处理
准确称取经组织粉碎机粉碎的豆芽样品2.00 g于50 mL塑料离心管中,加入10 mL 0.1%冰醋酸-乙腈溶液,涡旋混匀,超声15 min,加入1.5 g NaCl,涡旋混匀,以8 000 r/min离心3 min,取上清液于10 mL玻璃管中,氮气吹干,用甲醇-水(体积比3∶7)定容至1 mL,加入40 mg C18和40 mg PSA,涡旋混匀,过0.22 μm有机滤膜后于进样小瓶中,供LC-MS/MS测定。
1.4 仪器条件
1.4.1 色谱条件 色谱柱:Waters Atlantis T3色谱柱(150 mm×2.1 mm,5 μm)。流动相:A为1 mmol/L醋酸铵,B为甲醇,采用梯度洗脱方式;洗脱程序:0~1.0 min,10%B;1.0~4.0 min,10%~90%B ;4.0~7.0 min,90%B;7.0~7.1 min,90%~10%B;7.1~9.0 min,10%B。流速:250 μL/min;进样量:25 μL;柱温:室温。
1.4.2 质谱条件 离子源:电喷雾离子源(ESI);扫描方式:正负离子分段扫描;监测模式:选择离子反应监测(SRM);毛细管温度:350 ℃;鞘气流速:16 L/min;辅助气流速:2 L/min;扫描宽度:0.01;扫描时间:0.01 s;Q1分辨率:0.7 Da;Q3分辨率:0.7 Da。8种化合物的定性定量离子对及其碰撞能量见表1。
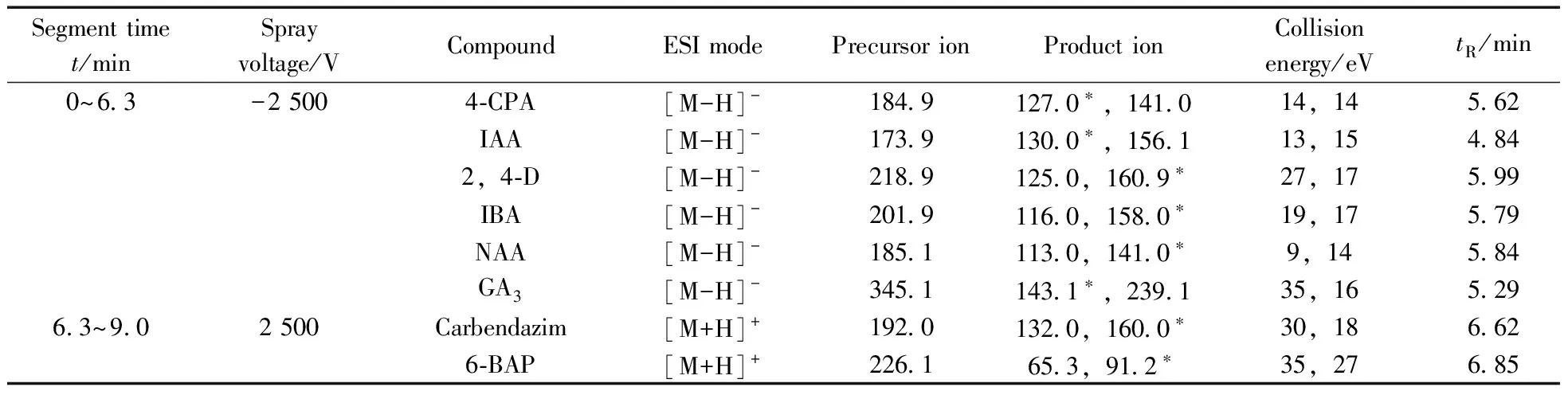
表1 8种化合物的LC-MS/MS 选择反应监测(SRM)参数
* quantitation ion
2 结果与讨论
2.1 样品的提取
根据豆芽基质的特点,本文采用QuEChERs方法,从提取方式、提取溶剂的选择、盐的选择和用量、分散固相萃取剂的选择和用量4个方面对黄豆芽和绿豆芽样品的测定条件进行优化,并以2,4-D和多菌灵作为代表,以考察不同测定条件对待测物回收率的影响。
2.1.1 提取方式 比较了振荡提取、超声提取两种最常用的提取方式对黄豆芽和绿豆芽中2,4-D和多菌灵回收率的影响。准确称取(2.00±0.05) g样品,加入10 mL 0.1%(体积分数)冰醋酸-乙腈,分别采取超声(15 min)和振荡(40 min)提取,每种基质每种提取方式做两个平行。结果显示,采用超声提取时,2,4-D和多菌灵的回收率比振荡提取时高,且提取时间明显少于振荡提取。因此,实验选择超声提取方式对豆芽中8种药物进行提取。
2.1.2 提取溶剂的选择 待测8种药物的化学性质差异较大,在不同溶剂中的溶解度不同。因此,本研究根据这8种药物的性质选取0.1%冰醋酸-乙腈、乙腈、丙酮、乙酸乙酯4种有机溶剂进行考察。准确称取(2.00±0.05) g样品,加入10 mL提取溶剂,超声15 min,每种基质、每种提取溶剂做两个平行。结果显示,以0.1%冰醋酸-乙腈作为提取溶剂时,两种基质(黄豆芽、绿豆芽)中2,4-D和多菌灵的回收率最高(见表2)。因此,实验选择0.1%冰醋酸-乙腈作为最佳提取溶剂。

表2 不同提取溶剂对豆芽中2,4-D和多菌灵回收率的影响
2.1.3 盐的选择及用量 在QuEChERS方法中一般会加入一定量的无水硫酸镁、无水乙酸钠和氯化钠,以去除水分和水溶性杂质,促进待测物向乙腈溶液中转移。本实验发现,加入无水硫酸镁和无水乙酸钠后,2,4-D的回收率升高至85%,但多菌灵的回收率降至26%。说明无水硫酸镁和无水乙酸钠的加入能够促进2,4-D提取,但对多菌灵有很强的吸附作用。而加入氯化钠时,8种待测物的吸附作用很小,甚至无吸附作用。因此,本研究选用NaCl对样品进行除杂净化,考察了不同的NaCl用量(0.5,1.0,1.5,2.0 g)时豆芽中2,4-D和多菌灵的回收率,结果见表3。结果表明,当NaCl的加入量由0.5 g增加到1.5 g时,2,4-D和多菌灵的回收率明显提高;但当NaCl的加入量超过1.5 g时,2,4-D和多菌灵的回收率反而降低。因此,实验选择加入1.5 g NaCl。
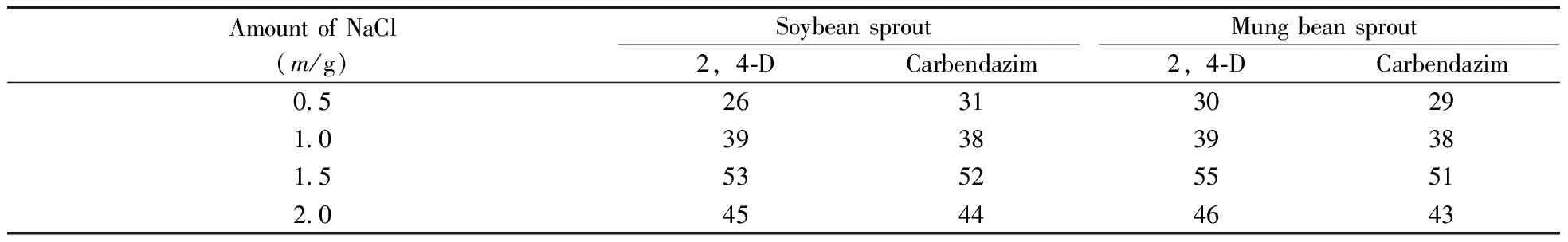
表3 不同用量的NaCl对豆芽中2,4-D和多菌灵回收率的影响
2.1.4 分散固相萃取剂的选择及用量 QuEChERS方法常会加入一定量的分散固相萃取剂(如十八烷基硅烷(C18)、N-丙基乙二胺(PSA)和石墨化碳黑(GCB)),以达到除杂净化的作用。C18可去除基质中的脂肪等弱极性化合物,PSA可去除基质中的有机酸、脂肪酸等极性化合物,GCB可吸附色素等物质。根据豆芽基质的特点,实验分别考察了3种分散固相萃取剂的加入量对豆芽中2,4-D及多菌灵回收率的影响(见表4)。结果显示:当C18的加入量由20 mg增至40 mg时,两种基质中2,4-D和多菌灵的回收率显著提高,说明C18的加入有较强的净化作用,明显减弱了基质抑制效应;但当C18的加入量达到60 mg时,2,4-D和多菌灵的回收率明显降低,说明过量的C18加入可能存在一定的吸附作用。当PSA的加入量由20 mg增至40 mg时,两种基质中2,4-D和多菌灵的回收率显著提高,说明PSA的加入有明显的净化作用,显著降低了基质抑制效应;但当PSA的加入量达到60 mg时,2,4-D和多菌灵的回收率显著降低,说明PSA过量加入可能导致吸附作用。随着GCB用量的增多,2,4-D和多菌灵的回收率明显降低,说明GCB对其有很强的吸附作用。
由于单独加入40 mg C18或40 mg PSA时,2,4-D和多菌灵的回收率均显著提高。为进一步确认分散固相萃取剂的种类和用量,本研究考察了同时加入40 mg C18和40 mg PSA对8种药物回收率的影响。准确称取(2.00±0.05) g样品,加入10 mL 0.1%冰醋酸(体积分数)-乙腈,超声提取,加入1.5 g NaCl后涡旋混匀,高速离心,取上清液氮气吹干,用甲醇-水(体积比3∶7)定容至1 mL,同时加入40 mg C18和40 mg PSA。结果显示,与单独加入C18或PSA时相比,8种药物的回收率均明显提高,达到78%~89%。说明同时加入40 mg C18和40 mg PSA后,净化作用明显增强,基质抑制效应显著降低。因此,实验选择同时加入40 mg C18和40 mg PSA。

表4 不同加入量的分散固相萃取剂对豆芽中2,4-D和多菌灵回收率的影响
2.2 仪器条件的优化
2.2.1 质谱条件的优化 由于4-氯苯氧乙酸、1-萘乙酸、2,4-二氯苯氧乙酸、吲哚乙酸、吲哚丁酸和赤霉素的分子结构中均含有羧基,因此采用负离子方式对其进行电离;而多菌灵和6-苄基腺嘌呤的分子结构中均含有氨基,因此采用正离子模式对其进行电离。质谱条件见表1。
2.2.2 色谱条件的优化 本研究选择Waters Atlantis T3柱分离8种化合物,以甲醇作为有机相进行梯度洗脱。由于分析物多为酸性化合物,流动相的pH值会对其解离产生一定影响,从而影响其离子化效率和保留行为。因此,本研究考察了甲酸、水和醋酸铵分别作为水相时对8种化合物分离效果的影响。结果表明,在甲酸溶液中,负离子的形成受到抑制,其中吲哚乙酸和吲哚丁酸的抑制程度最强;在水溶液中,吲哚乙酸的保留较弱,在2 min前出峰。因此,本研究采用醋酸铵溶液作为水相,并进一步考察了其浓度不同(0.05,0.5,1.0,5.0 mmol/L)时对8种化合物分离效果的影响。结果显示:当醋酸铵浓度分别为0.05 mmol/L和0.5 mmol/L时,吲哚乙酸的保留均较弱,在3 min前出峰;其他7种化合物的保留较好。当醋酸铵浓度为5.0 mmol/L时,萘乙酸的响应低,达不到检测要求;当醋酸铵浓度为1.0 mmol/L时,各待测物的响应、峰形、保留均较好,达到最佳检测效果。因此,实验选择1.0 mmol/L醋酸铵-甲醇作为流动相。
2.3 线性关系与定量下限
在优化实验条件下,对一系列标准工作溶液进行测定。以标准品的峰面积(Y)为纵坐标,对应质量浓度(X,μg/L)为横坐标,绘制标准曲线。结果显示,8种药物在5~100 μg/L范围内呈良好的线性关系,相关系数(r2)不低于0.991 1(见表5)。以信噪比等于10确定方法定量下限,分别通过阴性黄豆芽和阴性绿豆芽加标测定,8种药物的定量下限均为5 μg/kg。

表5 8种化合物的回归方程、相关系数、加标回收率和相对标准偏差(n=6)
2.4 回收率与相对标准偏差
本研究选择5,10,20 μg/kg 3个浓度水平分别对黄豆芽和绿豆芽进行加标回收实验,各化合物的回收率及相对标准偏差见表5。黄豆芽中8种药物的加标回收率为71.6%~87.9%,相对标准偏差(RSD)不大于14.6%。绿豆芽中8种药物的加标回收率为73.7%~85.1%,相对标准偏差(RSD)不大于14.0%。可见,此方法适用于豆芽中8种药物的检测,其准确度及精密度满足检测要求。图1为黄豆芽中添加8种药物(10 μg/kg)的色谱图。








图1 黄豆芽中添加8种药物(10 μg/kg)的SRM色谱图
2.5 实际样品的检测
采用本文建立的方法,对市售843批豆芽样品进行检测。其中,485批豆芽中检出4-CPA,残留量范围为0.02~12.3 mg/kg;442批豆芽中检出6-BAP,残留量范围为0.03~11.4 mg/kg;82批豆芽中检出GA3,残留量范围为0.01~0.15 mg/kg;65批豆芽中检出2,4-D,残留量范围为0.01~0.23 mg/kg;其他化合物均有检出,但残留量较低。
3 结 论
本文建立了高效液相色谱-串联质谱法测定豆芽中8种药物的分析方法。该法的选择性好、灵敏度高、抗干扰能力强,从而降低了对样品前处理的净化要求,提高了提取效率。同时通过优化前处理,基质抑制现象得到消除,方法的回收率满足检测要求。该方法可快速检测豆芽中8种药物的残留量,适用于市场上豆芽的质量监测。
[1] Liu H,Wu B,Yin Y,Xu W,Gui Q W,Yu K Y,Gong Y X,Zhao Z Y,Lin H,Shen W J,Shen C Y,Zhang R.Chin.J.Chromatogr.(柳菡,吴斌,殷耀,许蔚,桂茜雯,余可垚,龚玉霞,赵增运,林宏,沈伟健,沈崇钰,张睿.色谱),2013,31(1):22-26.
[2] Office of the Ministry of Health.The Related Reply on Standards for Uses of Food Additives(GB2760-2011),China Food Additives(卫生部办公厅.食品添加剂使用标准(GB2760-2011) 有关问题的复函,中国食品添加剂).
[3] GB2760-2011.National Food Safety Standards-Standards for Uses of Food Additives(GB2760-2011.食品安全国家标准食品添加剂使用标准).
[4] Li X P,Chen X H,Yao X P,Jin M C,Jiang J W.Chin.J.HealthLab.Technol.(李小平,陈晓红,姚浔平,金米聪,蒋经纬.中国卫生检验杂志),2005,15(2):149-150.
[5] Li X P,Jiang J W,Fan J Z,Jin M C,Yao X P.Chin.J.HealthLab.Technol.(李小平,蒋经纬,范建中,金米聪,姚浔平.中国卫生检验杂志),2006,16(3):267-269.
[6] Huang W P.Chin.J.Prevent.Med.(黄卫平.中华预防医学杂志),2002,36(1):44-45.
[7] Xia H,Lu Z Q,Yang Y Z.J.YangzhouUniv.:Agric.LifeSci.Ed.(夏慧,陆自强,杨益众.扬州大学学报:农业与生命科学版),2011,32(4):86-89.
[8] Wang Y Q,Zhang G H,Zhao X Z,He H J.FoodSci.Technol.(王一茜,张广华,赵学志,何洪巨.食品科技),2013,38(10):316-319.
[9] Chen J,An D G,Xu L,Gao L J,Xiao H Z,Chen Z,Li X L.ChemistryOnline(陈君,安东各,许莉,高连杰,肖宏展,陈致,李喜来.化学通报),2014,77(9):916-918.
[10] Min J C,Chen S J.Chin.J.Mod.Agric.Sci.Technol.(闵季春,陈石金.现代农业科技),2014,(10):282-284.
[11] Hu X N,Gu Y P,Lin H C,Wang R,Fang C F.Chin.J.FoodSci.(胡祥娜,顾亚萍,林惠纯,王瑞,方长发.食品科学),2014,35(20):253-257.
[12] Zhang J W,Guo C H,Ge S H,Zhang H C,Wang J,Dou C H,Ai L F.Chin.J.FoodHyg.(张静雯,郭春海,葛世辉,张海超,王敬,窦彩虹,艾连峰.中国食品卫生杂志),2014,26(5):441-445.
[13] Liu C S,Luo H Y,Xian Y P,Wang B,Wang L,Luo D H,Guo X D.J.Chin.MassSpectrom.Soc.(刘春生,罗海英,冼燕萍,王斌,王莉,罗东辉,郭新东.质谱学报),2014,35(4):303-310.
[14] Jin M C,Ren Y P,Chen X H.Chromatographia,2007,66:407-410.
[15] Chang Y W,Wu X Z,Li W,Hao L H.Sci.Technol.FoodInd.(常宇文,吴晓宗,李伟,郝莉花.食品工业科技),2007,28(12):203-205.
[16] Li Y,Xu X,Yuan H F,Gao H W,Feng H G.J.AnhuiAgric.Sci.(李殷,徐霞,袁荷芳,高蕙文,冯华刚.安徽农业科学),2014,42(27):9527-9528.
[17] Wang C Y,Liu J T,Gao L N,Zhu J,Song Y.TheLearningProblemsinPublicSecurityResearchProgress(the Third Volume)(王春媛,刘俊亭,高丽娜,祝娟,宋洋.公共安全中的化学问题研究进展.第三卷),2013:383-386.
[18] Yan J L,Yan Y Q,Wang L,Shi J W.Chin.J.HealthLab.Technol.(颜金良,颜勇卿,王立,施家威.中国卫生检验杂志),2006,16(10):1207-1208.
[19] Ding Y C,Jiang A X,Zhong M W.Chin.J.PublicHealth(丁友昌,姜爱香,钟鸣文.中国公共卫生),1997,13(2):77-77.
[20] Xi Z J,Zhang Z J,Sun Y H,Shi Z L,Tian W.Talanta,2009,79:216-221.
[21] Lehotay S J.MethodsBiotechnol.,2006,19:239-260.
DSC助力我国聚丙烯材料性能扩展
在现代高分子材料学发展中,树脂行业的发展尤为迅速,其中聚丙烯(PP)是热塑性树脂中增长速度最快的品种之一,其较高的性价比也使之成为商家竞相追逐的焦点。通过一定的技术手段,还可以赋予其更多的优异性能。譬如调控PP的晶型就是PP改性的重要手段之一。
中国科学院化学研究所的科研人员最近通过一定的技术手段合成了具有β-定向结晶特性的PP树脂,其中β晶含量占80%左右,且结构稳定。
该研究组利用DSC(示差扫描量热法)方法对β-定向结晶PP树脂的结晶动力学做了进一步探究,并与普通PP树脂的结晶行为进行了对比,得出其结晶速率的优越性。进而从结晶过程的微观角度对釜内聚合获得β-定向结晶PP树脂的方法进行了评价。
β晶型PP不仅具有普通树脂材料的基本性能,还具有较高的冲击强度、热变形温度以及加工延展性。基于这些优良特性,β晶型PP作为抗冲工程塑料制品及抗高温热变形制品在建筑、包装、家电及高速交通等行业都具有广泛的应用前景。
(信息来源:仪器信息网)
Simultaneous Determination of Eight Kinds of Drug Residues in Sprouts by High Performance Liquid Chromatography-Tandem Mass Spectrometry
ZHANG Ya-lian1,LIU Han2,WANG Sui-lou1*,DING Tao2*,XU Niu-sheng3,YU Ke-yao2,SHEN Wei-jian2,ZHAO Zeng-yun2,LIU Yun2,WU Bin2,ZHANG Rui2,SHEN Chong-yu2
(1.College of Pharmacy,China Pharmaceutical University,Nanjing 210009,China;2.Jiangsu Import & Export Inspection and Quarantine Bureau,Nanjing 210001,China;3.Life Science and Mass Spectrometry,Thermofisher Scientific,Shanghai 201206,China)
A high performance liquid chromatography-tandem mass spectrometric(LC-MS/MS) method was developed for the determination of eight kinds of drugs(4-chlorophenoxyacetic acid,indole-3-acetic acid,2,4-dichlorophenoxyacetic acid,indole-3-butyric acid,1-naphthlcetic acid,gibberellin acid,6-benzylaminopurine and carbendazim) in sprouts.The samples were extracted with acetonitrile containing 0.1% acetic acid,concentrated and cleaned up with dispersive solid phase extraction powder.The extract was analyzed by LC-MS/MS.The linear ranges of eight compounds ranged from 5 μg/L to 100 μg/L with correlation coefficients(r2) more than 0.99.The limits of quantitation of eight compounds were 5 μg/kg.At three spiked concentration levels of 5,10,20 μg/kg,the recoveries of eight compounds ranged from 71.6% to 87.9% with relative standard deviations(RSDs) not more than 14.6%.The method was accurate,simple and quick,and was suitable for the simultaneous determination of eight compound residues in sprouts.
high performance liquid chromatography-tandem mass spectrometry(LC-MS/MS);sprouts;drug residues
2014-10-31;
2014-12-10
国家科技支撑计划课题(2014BAK19B02,2012BAK08B01,2015KJ29)
10.3969/j.issn.1004-4957.2015.02.007
O657.63;TQ460.72
A
1004-4957(2015)02-0164-07
*通讯作者:王岁楼,博士,教授,研究方向:食品质量与安全,Tel:13912990382,E-mail:cpuwsl@126.com 丁 涛,硕士,高级工程师,研究方向:食品安全,Tel:025-52345193,E-mail:dingt@jsciq.gov.cn
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
食品安全导刊(2021年21期)2021-08-30
中老年保健(2021年4期)2021-08-22
昆明医科大学学报(2020年12期)2021-01-26
理化检验-化学分册(2020年12期)2021-01-26
基层中医药(2020年7期)2020-09-11
山东化工(2019年11期)2019-06-26
国际呼吸杂志(2019年1期)2019-03-08
学苑创造·A版(2017年10期)2017-12-21
核科学与工程(2015年2期)2015-09-26
