
超声提取-气相色谱质谱法测定大气PM2.5中正构烷烃
2015-04-26 00:57刘保献王小菊沈秀娥赵红帅张大伟
中国环境监测 2015年6期
刘保献,王小菊,常 淼,沈秀娥,赵红帅,张大伟
北京市环境保护监测中心,北京 100048
PM2.5对全球气候变化[1-4]、人体健康[5-8]、大气能见度[9-10]、大气化学[11-12]等均具有较大的影响。研究发现PM2.5中正构烷烃是常见的痕量有机污染物,主要来源于化石燃料燃烧[13]、高等植物排放[14]等。因其活性和挥发性低,其组成、分布、碳主峰、碳优势指数(CPI)等信息可以较好的反应污染源特征,使其成为气溶胶迁移和颗粒物来源研究中重要的标识物之一[15-16]。于娜[17]、牛红云[18]、胡冬梅[15]等对北京、南京、太原地区颗粒物中正构烷烃研究均发现,颗粒物中正构烷烃的浓度处于较高的水平。
目前正构烷烃的分析技术主要有气相色谱法[19]、气相色谱-质谱法[20]等,前处理主要方法有超声提取法[17]、手动索氏提取法[21]、热解析法[22]等。目前对正构烷烃分析监测方法的研究主要以地矿领域为主,大气PM2.5中正构烷烃的分析测试技术研究较少。而随着大气PM2.5组成特征及来源解析研究的深入,正构烷烃较好的示踪性质及独特的碳优势指数等[23]在PM2.5来源分析等方面具有很好的应用价值[24],越来越受到关注。
本文使用气相色谱质谱法测定大气PM2.5中C9~C40的正构烷烃,优化了测定仪器的色谱条件、离子源温度等条件,比较了不同的前处理方式对方法性能的影响,研究了方法的检出限、精密度、准确度、回收率等性能指标,同时结合实际样品测定进一步对方法的适用性进行确定。
1 实验部分
1.1 试剂与材料
C9~C40正构烷烃标准样品:(500.0 μg/mL,美国Accustandard)DRH-008S-R2;内标:六甲基苯,99.5%,德国 Dr.Ehrenstorfer;替代物:十四烷-d30,98%,美国 CIL;二十四烷-d50,98%,美国 CIL,DLM-2209-05;三十六烷-d74,98%,加拿大 CDN,A138P40;滤膜:美国 Whatman,石英,47 cm。
有机溶剂:二氯甲烷,农残级,J.T Baker;正己烷,农残级,J.T Baker;微量注射器(10.0 μL、100.0 μL、1.0 mL)。
1.2 仪器与设备
Agilent-7890B/5977(美国)气相色谱质谱仪,EI源;KQ-700CDE超声提取仪(45000 HZ、700 w);Filt-ex-12聚四氟过滤器(12位,意大利);DryVap定量浓缩仪(6位,美国);Agilent-7696A样品制备平台(美国);TH-16A型四通道PM2.5采样器(16.7 mL/min)。
1.3 实验方法
1.3.1 样品前处理
超声提取:将PM2.5样品膜放入洁净的厚壁直壁瓶内,使用100.0 μL的微量注射器,向PM2.5样品膜上加入100.0 μL的十四烷-d30替代物溶液(2.0 μg/mL),然后加入20 mL正己烷溶剂浸没样品膜。将提取瓶用内衬铝箔的螺纹盖密封后,放入超声仪中进行超声提取,超声萃取条件为超声功率45 khz,超声时间20 min完毕后,使用聚四氟过滤器将溶液过滤转移至收集瓶,继续向提取瓶内加入20 mL二氯甲烷,再重复超声提取两次,合并所有的提取溶液。每次超声后更换新水,保持较低的温度或加冰块降温。
手动索氏提取:将PM2.5样品膜装入索氏提取管中,使用100.0 μL的微量注射器,向PM2.5样品膜上加入100.0 μL的十四烷-d30替代物溶液(2.0 μg/mL),以 60 mL(约为提取烧瓶体积的三分之二)二氯甲烷-正己烷混合液(体积比为2∶1)浸泡4 h,调节提取温度为40~50℃,控制虹吸收率每小时6~10次,提取6 h。待提取完毕,使用定量浓缩仪,浓缩温度40℃,体系负压68 950 pa,正己烷换相并浓缩至0.9 mL,使用正己烷最终定容到1.0 mL,样品制备平台自动加入10.0 μL 内标物(六甲基苯,10.0 μg/mL),于4 ℃冰箱中存储待测。
1.3.2 GC-MS条件
色谱条件:DB-5MS(30 m×0.25 mm×0.25 μm)色谱柱,柱温:在50℃下保存10 min,然后以6℃/min的速率升温至300℃保存40 min,最后在320℃后运行2 min,进样口温度290℃,分流比5∶1,柱流量 0.8 mL/min,进样量 2.0 μL。
质谱条件:采用电子轰击电离方式(EI)进行离子化,EI电离能量为70 eV;质谱接口温度300℃;离子源温度300℃;四级杆温度150℃;溶剂延迟4.5 min。目标化合物、内标化合物及替代物保留时间、定量离子及定性离子见表1。
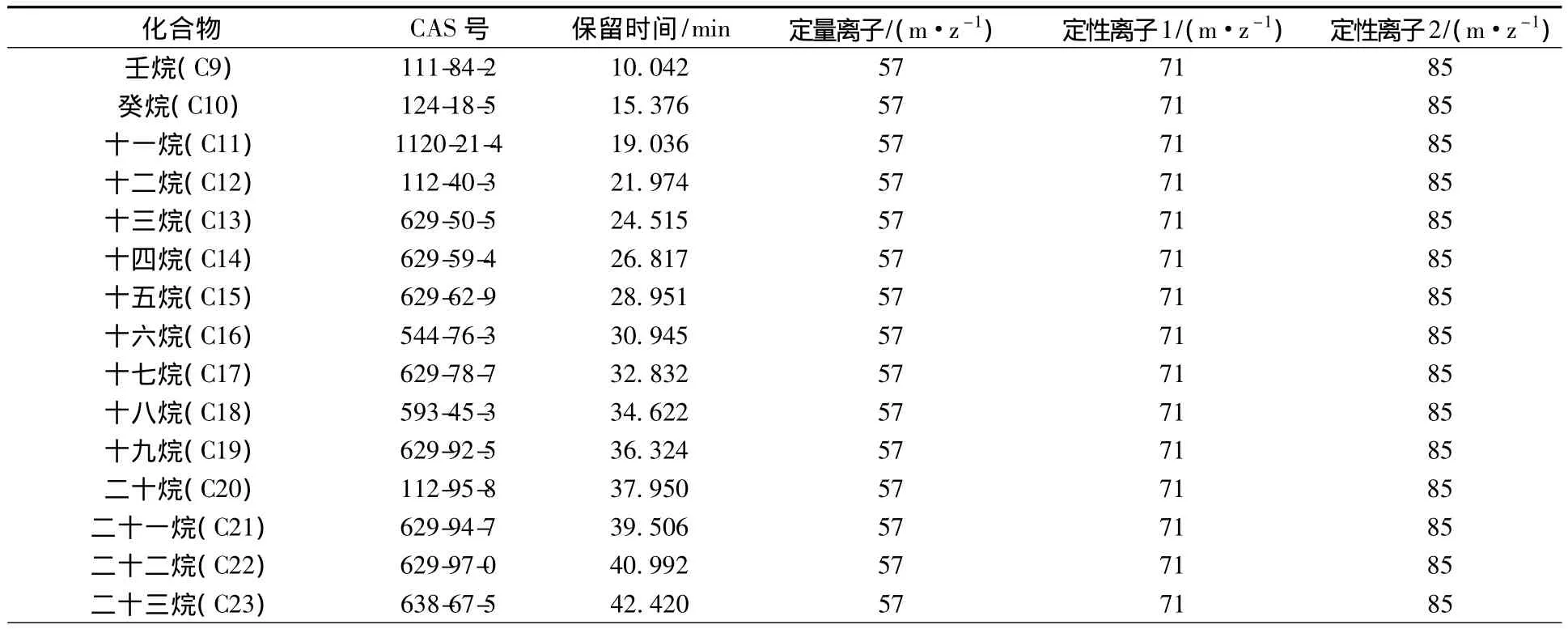
表1 目标化合物名称、色谱保留时间及质谱条件
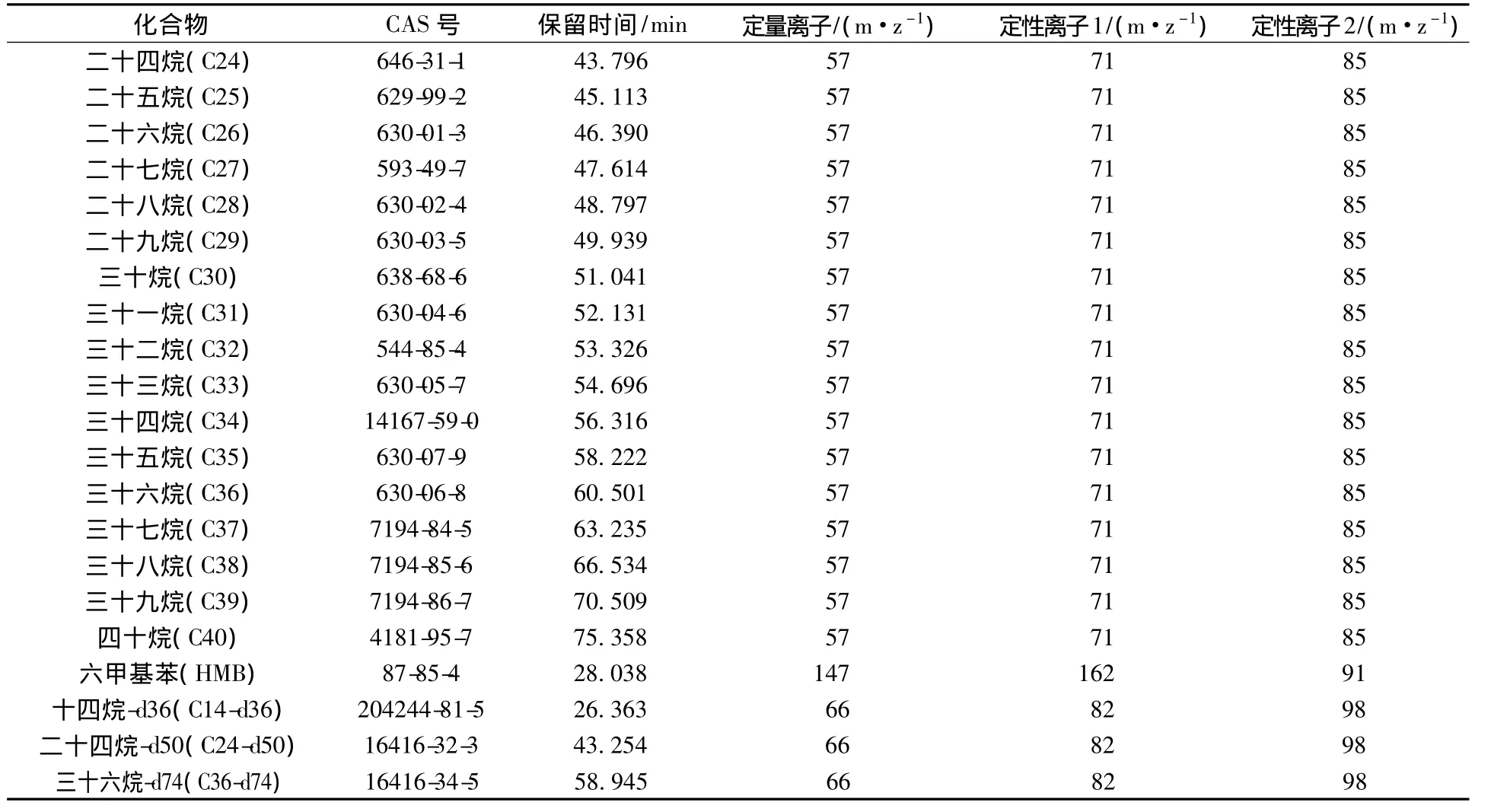
续表1
2 结果与讨论
2.1 色谱及质谱条件
2.1.1 色谱条件及分离
图1为 0.50 μg/mL的正构烷烃、0.196 μg/mL的六甲基苯及0.98 μg/mL的替代物的谱图。由图1可以看出,使用气相色谱质谱仪,在上述的色谱条件下(“1.3.2”小节所述)可以较好的分离32种C9~C40的正构烷烃、六甲基苯及3种替代物,化合物出峰尖锐,分离度较好,所选择的3种替代物可较均匀的分布在32种目标化合物之间,满足该方法的要求。同时,由质谱研究发现正构烷烃的的质谱图均为[CnH2n+1]+的碎片离子,出峰顺序按碳数基本等距出峰,且强度呈正态分布。
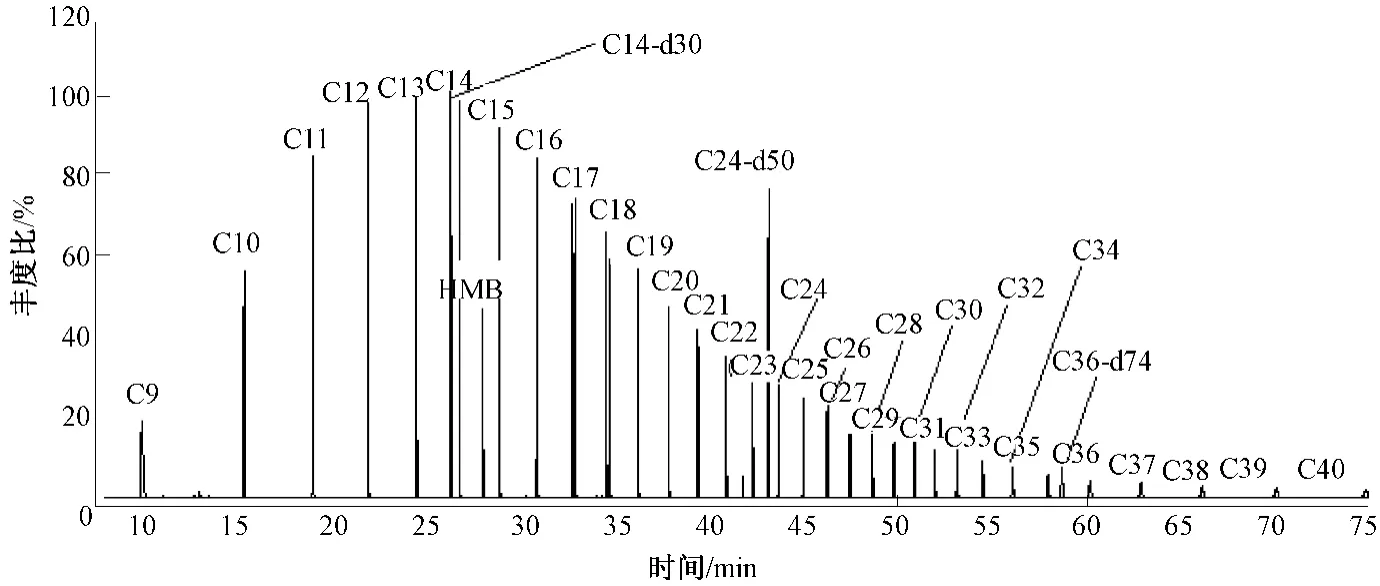
图1 32种正构烷烃、内标化合物及替代化合物的色谱图
2.1.2 离子源温度的选择
由图1可以看出,C9~C40各正构烷烃的峰强度呈明显的正态分布。其中高分子量的化合物响应逐渐降低,特别是C30以后的物质,分析检测难度加大。由于离子源温度的提高,一定程度可以加大离子化效率,并能保证离子源洁净度,防止高沸点物质残留。分别选择230、250、280、300℃离子源温度,查看32种正构烷烃的出峰效果,见图2。
由图2可以看出,离子源温度的增加可大大提高正构烷烃的灵敏度,32种化合物峰面积离子源温度的增加而增加,特别是C24烷烃以后的高分子量正构烷烃的增加幅度更为明显,其中离子源温度为300℃时C35烷烃的峰面积是230℃的4.6倍,可见离子源温度条件的优化对高分子量的正构烷烃测定具有重要的意义,因此选择300℃的离子源温度。
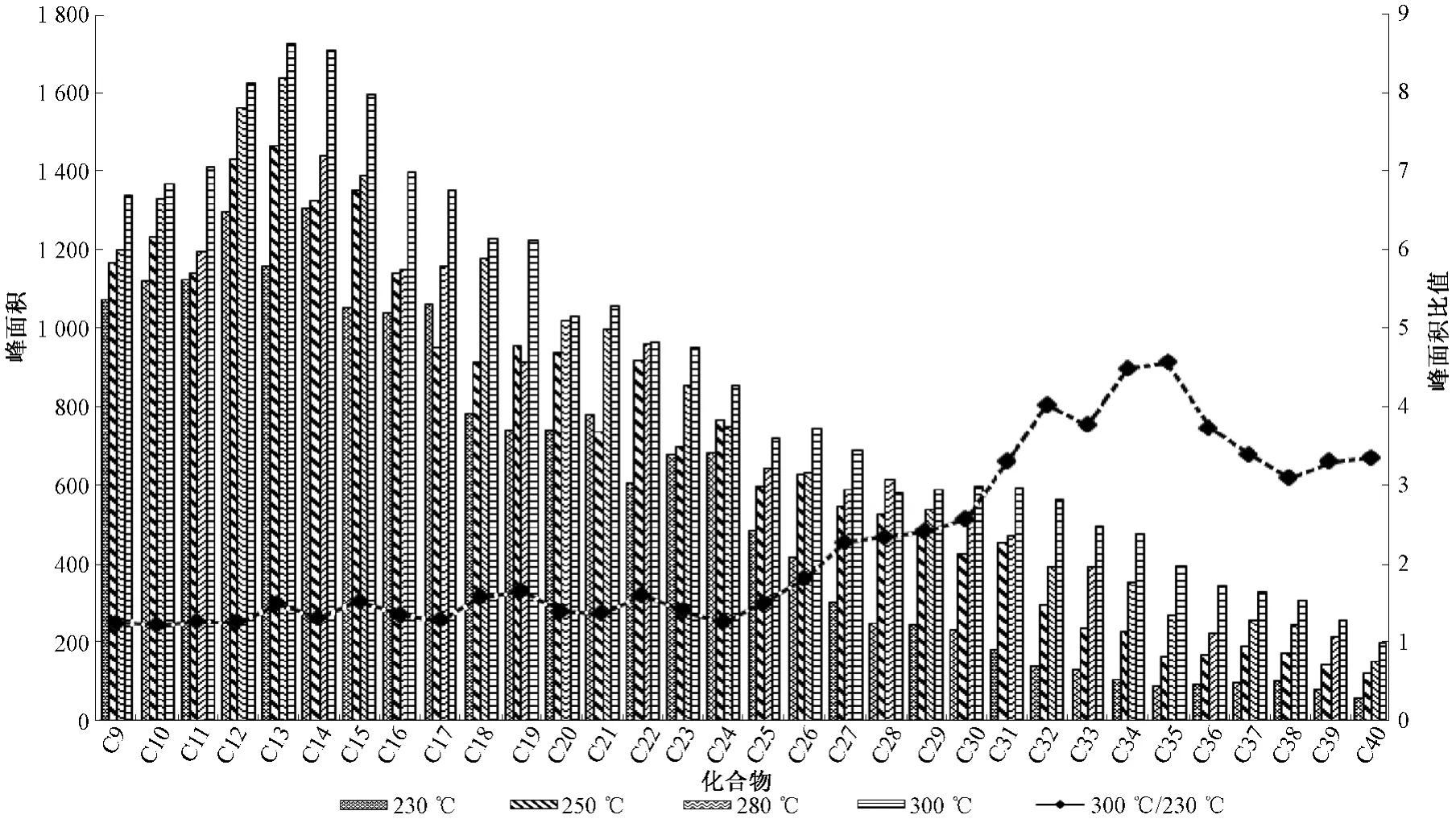
图2 不同离子源温度对峰面积的影响
2.2 方法前处理的比较
颗粒物中有机物化合物的前处理方法主要有超声提取法、手动索氏提取法等,为研究两种前处理对正构烷烃的提取效果,配置浓度分别为0.5、5.0、50.0 μg/mL的 正 构 烷 烃 标 准 溶 液,使用100.0 μL的微量注射器,分别向空白石英滤膜加入100.0 μL配制好的3种标准溶液,配制正构烷烃含量分别为50.0、500.0、5 000.0 ng 3种低、中、高浓度的空白加标样品,每种浓度空白加标样品分别使用超声提取、索氏提取进行前处理,比较两种前处理方法的加标回收率,结果见图3。
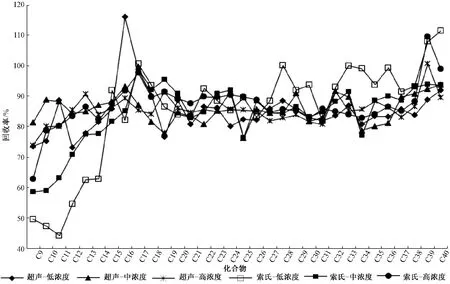
图3 不同前处理方式的考察
由图3可知,超声提取的3种浓度空白加标 回收率均为70% ~120%,而手动索氏提取对不同的化合物加标回收率相差较大。手动索氏提取在C17及以后的化合物略高于超声提取的加标回收率,但C17之前的化合物加标回收率明显较低,这可能是由于手动索氏提取时间较长,对高分子量、不易挥发的正构烷烃提取的效果更好;但对于相对易挥发的正构烷烃,长时间回流前处理会造成较多损失。考虑到全部化合物,方法宜选择超声提取进行颗粒物前处理。
2.3 空白滤膜的预处理
空白石英滤膜在生产、运输、保存等环节中往往易受外界的影响,吸附一定的有机物,使空白本底增加以致影响结果,特别是针对痕量有机化合物的监测。目前,常用去除有机物的方法为灼烧法,胡冬梅等[15]使用500℃,灼烧5 h去除空白石英滤膜上的正构烷烃本底。牛红玉等[18]则采用550℃,灼烧2 h。唐小玲等[25]使用450℃,灼烧4 h。为研究不同灼烧时间对空白石英滤膜正构烷烃本底去除效果的影响,分别在550℃的灼烧温度下,比较0、2、4、6 h不同灼烧时间对空白值的影响,见图4。
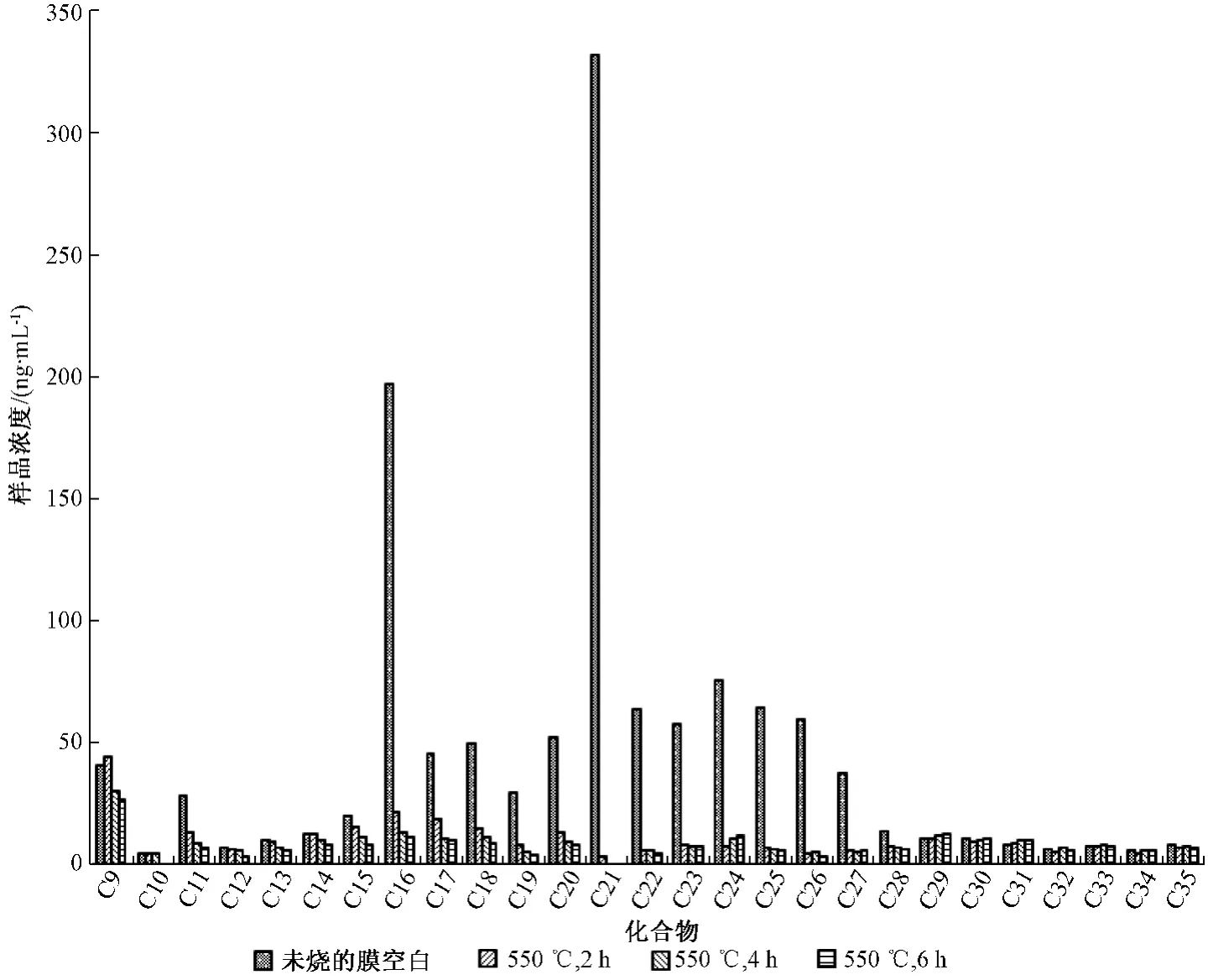
图4 不同灼烧时间对膜空白的影响
由图4可以看出,未进行灼烧的石英滤膜含有较高浓度的正构烷烃,浓度范围为0~332ng/mL,将大大影响正构烷烃的测定效果。在550℃温度下,分别经2、4、6 h灼烧后,空白值均能大大的降低,相比较C22烷烃以后的化合物3种不同的灼烧温度效果基本相当(C36~C40均未检出),C22之前的化合物与4、6 h的灼烧效果相差不大,略好于2 h。因此,本文选用空白石英滤膜预处理方式为550℃温度下,灼烧4 h。
2.4 方法性能指标
2.4.1 标准曲线及检出限
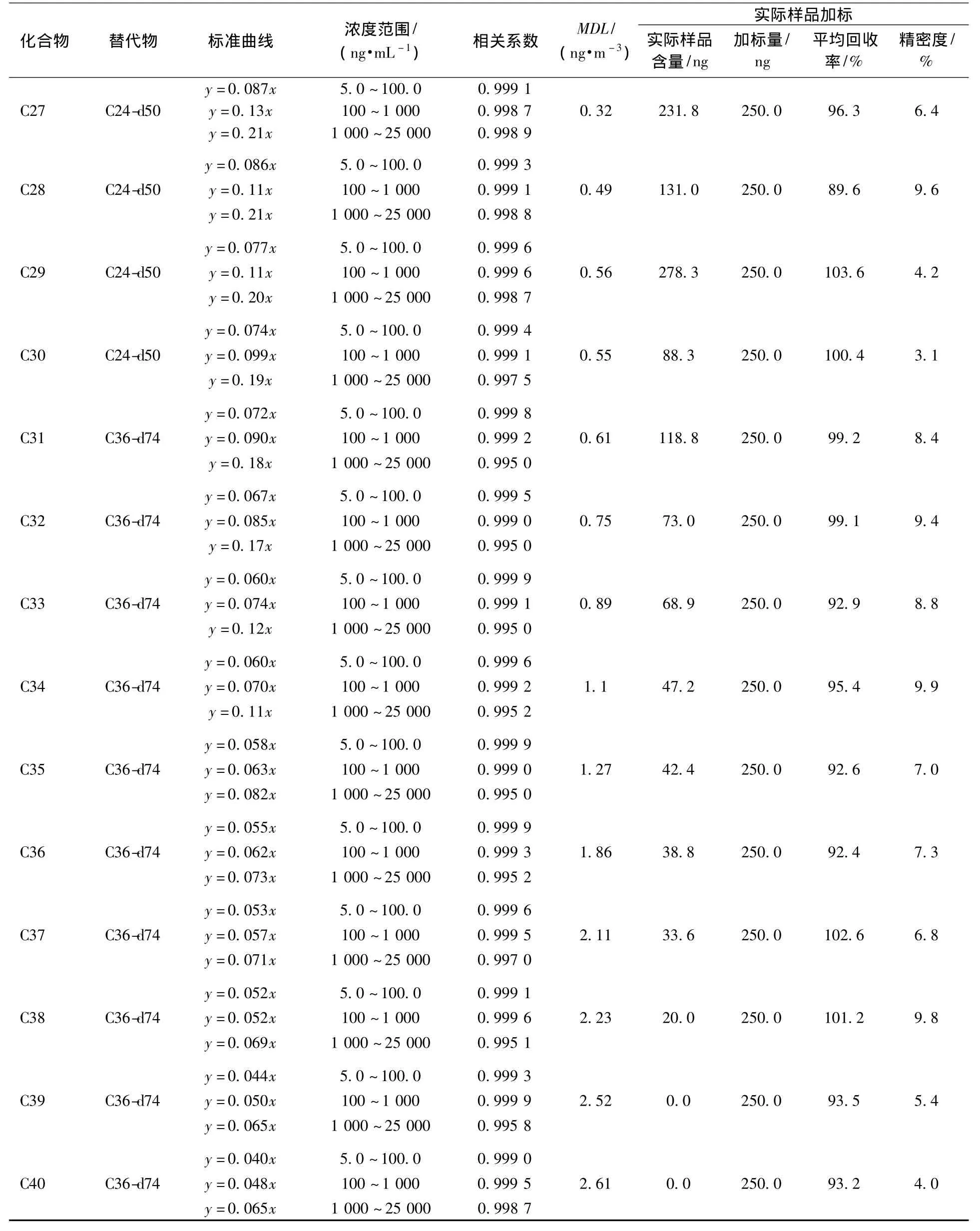
使用正构烷烃标准溶液及微量注射器,用正己烷作为溶剂,分别配置 25.0、10.0、5.0、2.5、1.0 μg/mL,1 000、750、500、250、100、100、50、25、10、5 ng/ml高、中、低3种不同浓度的标准曲线,分别加入 10.0 μL、20.0 μg/ml的内标液(六甲基苯),经仪器测定3种浓度的标准曲线的相关系数均在0.995以上,见表2。
按照标准[26]要求,连续测定7次5.0 ng/mL空白样品、50.0 ng/mL的空白加标样品,计算方法目标化合物的方法检出限(MDL),计算公式为MDL=3.143×S,式中,S为7次测定结果的标准偏差。按上述公式计算方法的检出限,由表2可得,在分流比5∶1的情况下,使用小流量采样器及47 cm的石英滤膜采集环境空气中的PM2.5,采样流速为16.7 L/min,采集时间为24 h,体积为24 m3时,各目标化合物的方法检出限在0.046~2.6 ng/m3,具有较好的灵敏度。
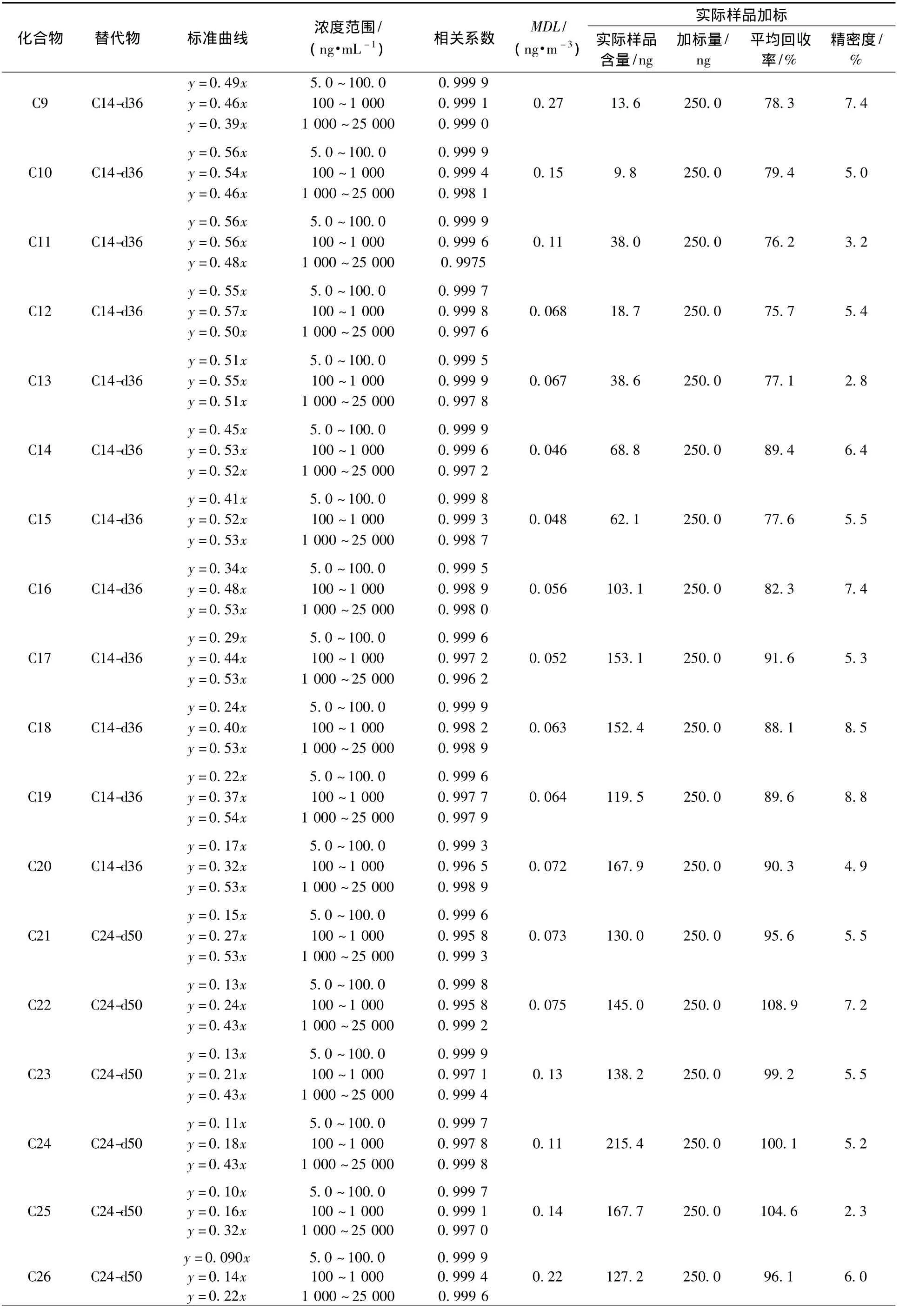
表2 方法标准曲线、检出限、精密度和准确度等性能指标
续表2
2.4.2 精密度和准确度
分别选择 50.0、500.0、5 000.0 ng 3 种高低不同浓度的目标化合物进行空白膜的加标实验,每种浓度重复6次,考察超声提取-气相色谱质谱法测定PM2.5中正构烷烃的加标回收率、精密度,该方法空白加标回收率较好,低、中、高3种浓度中各化合物平均加标回收率范围为72.2%~117.8%、73.5% ~104.4%、73.8% ~109.5%,精密度均小于10%。
为考察实际样品的加标回收率,使用小流量采样器(16.7 mL/min)、石英滤膜采集样品,在同一张PM2.5滤膜上裁取相同大小的滤膜4张,一张用于测定样品浓度,其余3张分别加入含量为250 ng的正构烷烃标准样品,按照上述前处理方法对样品进行处理,32种正构烷烃的加标回收率为75.7% ~108.9%(表2),和空白加标的回收率相差不大,3次测定的相对标准偏差也均低于10%,说明实际样品的基体干扰对本方法影响不大。
2.5 实际样品的测定
为考察方法的适用性,于2013年1、6月在北京城区车公庄点位(E116.32,N39.93)分别开展为期7 d的样品采集、分析,见图5。
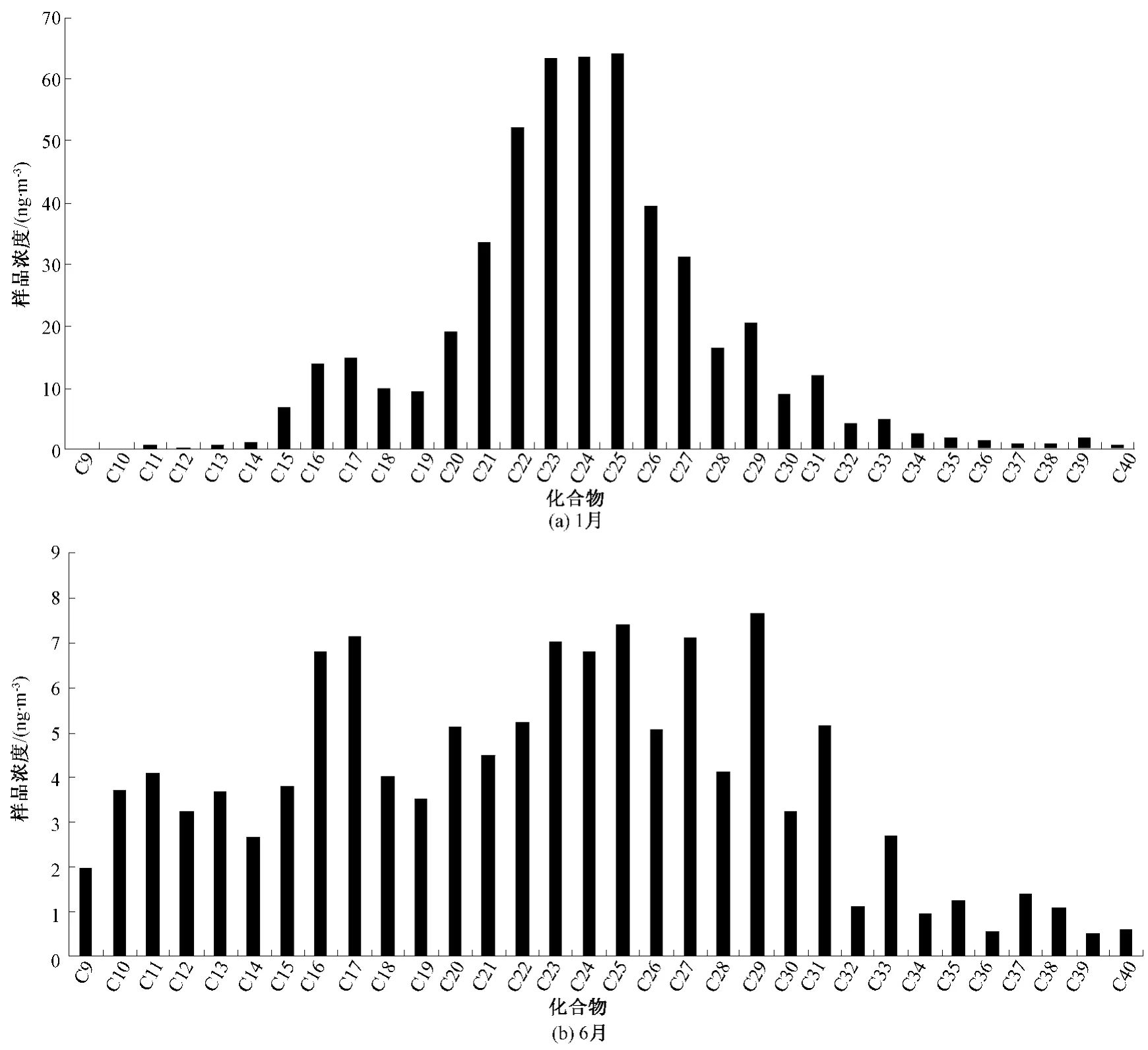
图5 北京城区1、6月实际样品中正构烷烃的测定
由图5可以看出,1月PM2.5中正构烷烃总量为506.9 ng/m3,范围为0.17~64.3 ng/m3;6月PM2.5中正构烷烃总量为 123.6 ng/m3,范围为0.53~7.67 ng/m3,各化合物均有检出。可见,即使在使用小流量采样器,该方法的检出限、测量范围也可较好的满足实际样品的测定。
3 结论
用超声提取-气相色谱质谱法测定大气PM2.5中32种正构烷烃,32种正构烷烃、内标化合物及3种替代物出峰尖锐,分离度较好,最佳的离子源温度为300℃,超声提取整体效果要优于手动索氏提取,石英滤膜采样前必须进行灼烧,最佳处理条件为处理方式为550℃温度下灼烧4 h。
该方法具有较好的性能指标,高、中、低3种浓度的标准曲线的相关系数均在0.995以上,3种浓度的空白膜样品加标回收率范围为72.2%~117.8%、73.5% ~104.4%、73.8% ~109.5%,精密度均小于10%,实际样品加标回收率为75.7%~108.9%,实际样品加标回收率和空白加标的回收率相差不大。当采样体积为24 m3时,各目标化合物的方法检出限在0.046~2.6 ng/m3。
通过2013年1、6月正构烷烃浓度范围分别为0.17~64.3、0.53~7.67 ng/m3的实际样品验证,使用本方法测定小流量采样器采集的PM2.5样品,各化合物均有检出,该方法的检出限、测量范围也可较好满足实际样品的测定。
[1]胡颖,邵龙义,沈蓉蓉,等.北京市PM2.5对DNA的氧化性损伤规律分析 [J].中国环境科学,2013,33(10):1 863-1 868.
[2]Hueglin C,Gehrig R,Baltensperger U,et al.Chemical characterisation of PM2.5,PM10and coarse particles at urban,near-city and rural sites in Switzerland [J].Atmospheric Environment,2005,39:637-651.
[3]Cao J,Chow J C,Lee F S C,et al.Evolution of PM2.5Measurements and Standards in the US and future perspectives for China[J].Aerosol and Air Quality Research,2013,13:1 197-1 211.
[4]Sun Y L,WangZ F,FuP Q,etal.Aerosol composition,sources and processes during wintertime in Beijing,China [J].Atmospheric Chemistry and Physics,2013,13:4 577-4 592.
[5]Yang D,Qi S,Devi N,et al.Characterization of polycyclic aromatic hydrocarbons in PM2.5and PM10in Tanggu District,Tianjin Binhai New Area,China[J].Frontiers of Earth Science,2012,6(3):324-330.
[6]Sun Z,Mu Y,Liu Y,et al.A comparison study on airborneparticles during haze days and non-haze days in Beijing[J].Science of the Total Environment,2013,456:1-8.
[7]Cao J,Xu H,Xu Q,et al.Fine particulate matter constituents and cardiopulmonary mortality in a heavily polluted Chinese city[J].Environmentalhealth perspectives,2012,120(3):373-378.
[8]郑永杰,刘佳,田景芝,等.齐齐哈尔市春季大气中PM2.5的污染特征分析[J].中国环境监测,2014,4(30):76-81.
[9]Chan Y C,Simpson R W,Mctainsh G H,et al.Characterization of chemical species in PM2.5and PM10aerosols in Brisbane Australia [J].Atmospheric Environment,1997,31:2 061-2 080.
[10]Chow J C,Watson J G,Lu Z,et al.Descriptive analysis of PM2.5and PM10at regionally representative locations during sjvaqs/auspex [J].Atmospheric Environment,1996,30:2 079-2 112.
[11]Cruz C N,Pandis S N.A study of the ability of pure secondary organic aerosol to act as cloud condensation nuclei[J].AtmosphericEnvironment,1997, 31:220-224.
[12]Malm W C . The effectsofmodelsofaerosol hygroscopicity on the apportionment of extinction[J].Atmospheric Environment,1997,31:1 965-1 976.
[13]王娟,钟宁宁,栾媛,等.鄂尔多斯市秋季大气PM2.5、PM10颗粒物中正构烷烃的组成分布与来源特征 [J]. 环 境 科 学 学 报,2007,27(11):1 915-1 923.
[14]Cecinato A,Marino F,Di Filippo P,et al.Distribution of nalkanes,polynuclear aromatic hydrocarbons and nitrated polynuclear aromatic hydrocarbons between the fine and coarse fractions of inhalable atmospheric particulates[J].Journal of Chromatography,1999,846(2):255-264.
[15]胡冬梅,彭林,白慧玲,等.太原市空气颗粒物中正构烷烃分布特征及来源解析[J].环境科学,2013,10(34):3 733-3 740.
[16]Gelencser A,Barcza T,Kiss G,et al.Distribution of n-alkanes and PAHs in atmospheric aerosols[J].Atmospheric Research,1998,46(3/4):223-231.
[17]于娜,魏永杰,胡敏,等.北京城区和郊区大气细粒子有机物污染特征及来源解析[J].环境科学学报,2009,2(29):243-250.
[18]牛红云,赵欣,戴朝霞,等.南京市大气气溶胶中颗粒物和正构烷烃特征及来源分析[J].环境污染与防治,2005,5(27):363-367.
[19]成玉,盛国英,闵育顺,等.珠江三角洲气溶胶中正构烷烃分布规律、来源及其时空变化[J].环境科学学报,1999,1(19):96-111.
[20]段凤魁,贺克斌.大气颗粒物中三类有机组分的萃取分离净化和GC/MS测定[J].质谱学报,2010,3(31):165-171.
[21]杜鹃.北京市海淀区大气PM10中正构烷烃的分布研究[J].安徽建筑工业学院学报:自然科学版,2007,6(15):32-36.
[22]刘浩,张勇,张家泉,等.黄石城区夏季大气微细颗粒物中正构烷烃污染状况及源解析[J].湖北理工学院学报,2013,3(29):26-29.
[23]Alves C,Pio C,Duart E A.Composit ion of ext ract abl e organic matter of air part icles from rural and u rban Portugu ese areas[J].Atmospheric Environment,2001,35(32):5 485-5 496.
[24]Huang X F,He L Y,Hu M,et al.Annual variation of particulate organic compounds in PM2.5in the urban atmosphere of Beijing[J].Atmospheric Environment,2006,40:2 449-2 458.
[25]唐小玲,毕新慧,陈颖,等.广州市空气颗粒物中烃类物质的粒径分布[J].地球化学,2005,34(5):508-514.
[26]HJ/T 168—2010 环境监测 分析方法标准制修订技术导则[S].
猜你喜欢
纺织标准与质量(2022年3期)2022-08-10
核技术(2022年7期)2022-07-22
金桥(2022年2期)2022-03-02
Plasma Science and Technology(2021年3期)2021-03-22
东坡赤壁诗词(2020年3期)2020-07-04
科技视界(2019年18期)2019-08-07
中国新技术新产品(2019年23期)2019-01-20
分析化学(2018年7期)2018-09-17
浙江大学学报(工学版)(2016年11期)2016-06-05
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
