
催化燃烧法脱除含氯挥发性有机化合物研究进展
2015-04-10 05:35:49王明玺刘善堂
化学工业与工程 2015年3期
王明玺,刘善堂
(武汉工程大学化学与环境工程学院,武汉 430074)
含氯挥发性有机物(Chlorinated volatile organic compounds, CVOCs),如二氯甲烷(CH2Cl2)、三氯乙稀(C2HCl3)、氯苯(C6H5Cl)、多氯二苯并二噁英(PCDDs)等是常见的工业化学试剂,多以工业废水和废气的形式排放而污染环境。绝大部分含氯挥发性有机物挥发性高,具有良好的化学和热稳定性,不易被分解或生物降解,可以在自然界中长期滞留,对环境造成持久性的污染;且有较高的毒性和致癌、致畸作用,是一类严重威胁人类和生物健康、破坏自然生态环境的化合物。因此,含氯挥发性有机物的去除已成为环境污染治理的焦点问题。
对CVOCs最好的治理是提高生产效率、减少排放,然而这在大多数情况下是不现实或不可能的。因为CVOCs应用非常广泛,是许多工业中不可替代的原料,且生产效率的提高是有限度的,故对CVOCs的后续治理十分关键和必要。对CVOCs的脱除治理采用的方法有直接焚烧[1]、水蒸气重整[2]、催化加氢脱氯[3]、光催化氧化[4]和催化燃烧法[5]等。以上几种方法中,前4种方法都存在一些明显的缺点,如直接焚烧法燃烧温度高(>1 000 ℃),且不完全燃烧会产生有害物质,需要大量的能量补给;水蒸气重整法为CVOCs被吸附在催化剂活性位上,然后与水蒸气反应而生成CO、H2和HCl,生成的HCL易使催化剂中毒而失活;催化脱氢加氯法需要储存大量的氢气和贵金属,而贵金属催化剂容易氯中毒严重且不易工业化;光催化氧化法尽管消除CVOCs具有很好的活性,但存在量子效率低、反应速度慢等缺点制约了光催化氧化技术在CVOC脱除上的实际应用。相比较而言,催化燃烧是近十多年来发展起来的一种新型处理CVOCs的技术,具有显著的优点如较低的能量需求(燃烧温度低)、成本低和不会形成NOx,被认为是最可行和最有希望的CVOCs脱除的方法,成为近年来研究的热点。
在众多的含氯挥发性有机物中,用于催化燃烧研究的化合物有:1,2-二氯乙烯(DCE)、三氯乙烯(TCE)、二氯甲烷(DCM)和氯苯(CB),其它的CVOCs如四氯乙烯、四氯化碳和氯仿偶有涉及。这些化合物的活性差别很大,通常情况下饱和含氯挥发性有机物比不饱和含氯挥发性有机物活性更高,其活性与它们的吸附容量有关。本论文将总结近年来用于催化燃烧含氯挥发性有机物的催化剂,讨论其活性、选择性和稳定性,并对催化剂催化燃烧目标物的机理进行综述。
1 催化燃烧脱除CVOCs催化剂
催化燃烧CVOCs是指在催化剂作用下,废气中的有害组分被氧化剂完全氧化,理想的最终产物为二氧化碳(CO2)和氯化氢(HCl)。其优点是耗能小、操作温度低、脱除效率高,低温下可以处理较低体积分数(<1 %)的污染物废气。选用合适的催化剂是催化燃烧脱除CVOCs的关键。目前,用于低温催化燃烧CVOCs的催化剂主要有贵金属催化剂、过渡金属氧化物、钙钛矿型氧化物催化剂、以及层柱状黏土和固体酸催化剂等。
1.1 贵金属催化剂
催化燃烧CVOCs用作催化剂的贵金属一般有铂(Pt)、钌(Ru)、金(Au)、铑(Rh)和钯(Pd)等, 它们对C—Cl、C—O、C—H和H—H键具有较高的活性,因此被广泛应用于CVOCs的催化燃烧。表1列出了近年来催化燃烧脱除CVOCs的贵金属催化剂。
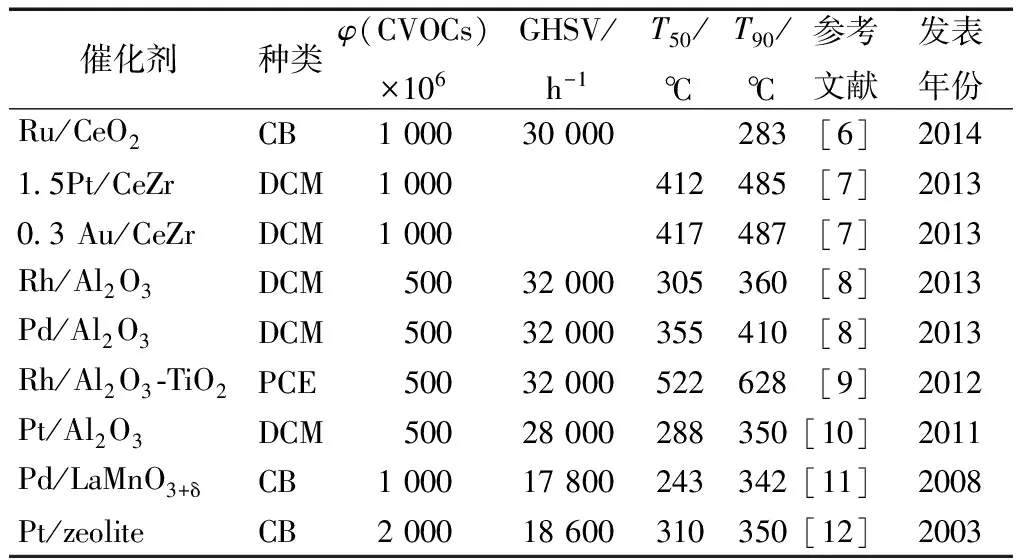
表1 催化燃烧 CVOCs 的典型贵金属催化剂
注:T50为CVOCs转化率为50%的温度,℃;T90为CVOCs转化率为90%的温度,℃; GHSV为空速;CB为氯苯;DCM为二氯甲烷;PCE为四氯乙烯。
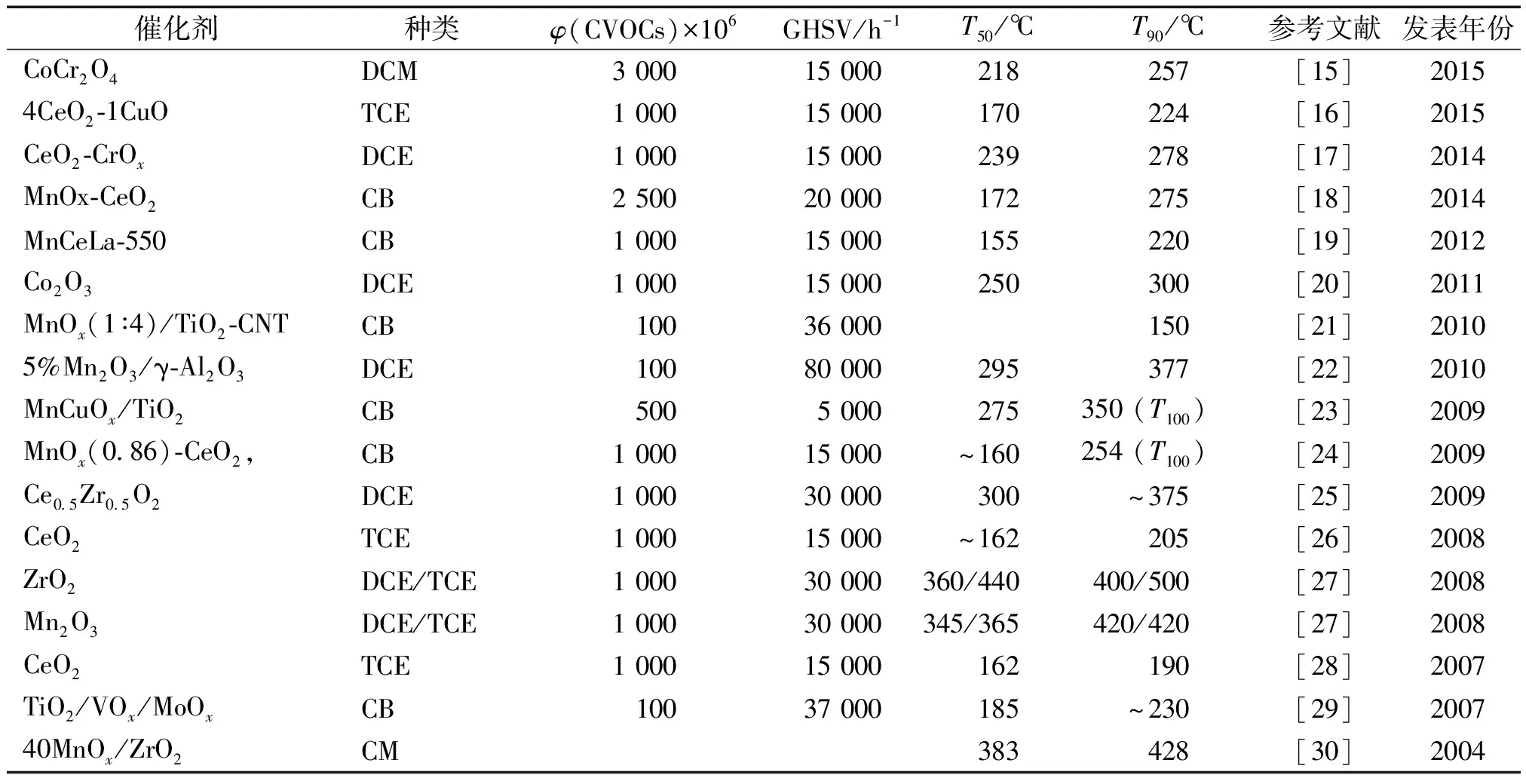
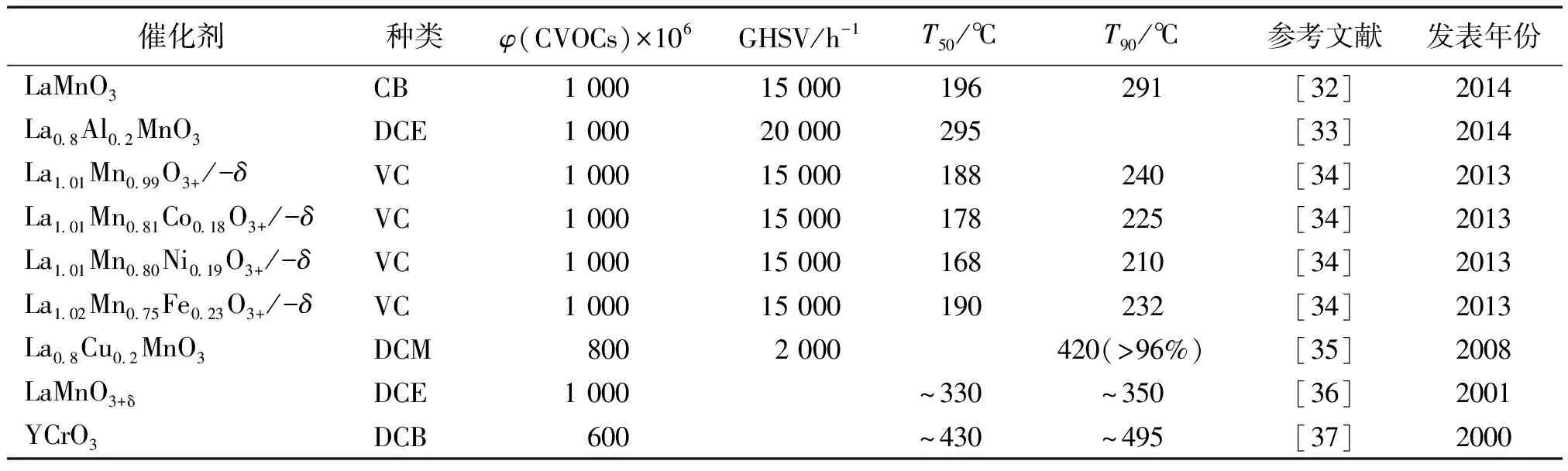
如表1所示,作为催化剂的贵金属一般是负载在载体上使用,这是因为贵金属价格昂贵载体的表面结构和化学性质对催化剂的性能有重要影响。作为载体的材料有Al2O3,SiO2,CeO2和沸石等通常具有较大的比表面积,以使贵金属能够充分地分散。Salvatore等[12]用等体积浸渍法制备了Pt负载型催化剂,选用γ-Al2O3、H-ZSM5和不同配比的SiO2/Al2O3为载体,用于催化燃烧2 000×106的氯苯(CB)。结果表明Pt/沸石比Pt/γ-Al2O3具有更高的催化活性,且多氯化苯(PhClx)的生成量顺序为:Pt/H-ZSM5 在贵金属催化剂中,Pd表现出了较高的催化燃烧CVOCs的活性[13], Pt能够促进含氯碳氢化合物完全氧化为CO2, 而Pd作为催化剂产生的CO含量更高。因为过量氧的存在,贵金属,特别是Pd容易被氧化和氯化,从而导致贵金属中毒[11,14]。除了易中毒外,贵金属催化剂成本高、氯代反应活性高即易产生更大的多氯副产物,在高温区容易因为贵金属的流失而导致失去活性等问题,因此使得贵金属的使用受到极大地限制,近年来得用贵金属作为催化剂的研究越来越少,开发更适宜的催化剂比如过渡金属氧化物、钙钛矿型催化剂等成为研究的热点。 过渡金属氧化物,如钒(V)、铬(Cr)、锰(Mn)、铁(Fe)、钴(Co)、镍(Ni)和铜(Cu)等,对CVOCs的催化燃烧具有良好的氧化活性和抗氯中毒能力,且资源丰富、价格低廉,是一类得到广泛研究的催化剂,在环境催化领域有很好的发展前景。如表2所示,列举了近十年来有代表性的过渡金属氧化物作为催化剂催化燃烧CVOCs的研究成果。 表2 催化燃烧CVOCs的典型过渡金属氧化物催化剂 注:T100为CVOCs完全转化时的温度。DCE为二氯乙烯;TCE为三氯乙烯;CM为氯甲烷。 从表2所列文献来看,过渡金属氧化物对CVOCs具有较高的降解活性,其中有些催化剂的活性甚至超过了贵金属催化剂,这得益于其高的电子迁移速率和正的氧化态。过渡金属氧化物既可以制成单一组分的催化剂,也可以制备复合金属氧化物,还可以负载在具有高比表面积的便宜的氧化物如Al2O3上制得负载型金属氧化物催化剂。在过渡金属化物催化剂中,氧化铬(Cr2O3)是活性最高的金属,因此对它的研究也最深入和广泛。氧化铬一般负载在Al2O3, 沸石、层柱状黏土、SnO2、TiO2、SiO2和多孔碳上。在催化燃烧TCE的过程中,负载2%的Cr/Al2O3的活性比2%Pd/Al2O3的活性更高。在铬催化剂表面,没有水存在的情况下,氧化过程中会生成少量的PCE,而由于不完全氧化生成的CO量较多。此外,低温下,铬会形成极毒的残余物,如铬酰氯而使其应用受到极大限制。 近年来,锰氧化物因其价格低廉、环境友好和高活性而引起广泛的兴趣,用于催化燃烧CVOCs研究的报道越来越多,如表2所示。锰氧化物是常见的过渡金属氧化物,可以形成多种价态的氧化物,如MnO、Mn3O4、Mn2O3和MnO2等,其中锰的价态与其负载量、载体种类、焙烧温度等制备方法、条件和锰的前驱物有关。在作为催化剂时,锰的最佳负载量决定于分散度、氧化态和氧的化学吸附能力。Ting等[22]用等体积浸渍法制备Mn2O3/γ-Al2O3用于二氯乙烯(DCE)的燃烧,转化率随初始温度和氧含量上升,而随着DCE的浓度和空速的增加而下降,且氧气和DCE浓度也会影响催化反应速率,氧化DCE的动力学行为满足幂指数定律。Mn2O3负载量为5%时,空速为80 000 h-1, 氧化浓度为100×106的二氯乙烯,295 ℃时其转化率为50%,当温度升至377 ℃时,转化率即达到90%。除了氧化锰外,二氧化铈(CeO2)也是一种高效的环境催化剂,因为其具有高的氧存储能力和Ce4+/Ce3+氧化还原对,CeO2和CeO2基复合金属氧化物廉价、环境友好、催化活性高,被大量应用于含氯有机物的催化分解。以硝酸铈为前驱物,采用热分解法制备的CeO2为催化剂催化燃烧三氯乙烯(TCE), CeO2展现出了很高的催化活性[26],在550 ℃锻烧的CeO2催化燃烧TCE时,完全燃烧TCE所需温度仅为205 ℃。CeO2的高活性源于其表面碱性、高的氧移动能力以及CeO2能够提供充足的氧。 纯CeO2作为催化剂,虽然活性很高,但因CVOCs分解产生的HCl或Cl2与CeO2有极强的吸附作用,从而堵塞活性位,将吸附在CeO2表面的氯或氯离子迅速移除或转移走,CeO2的稳定性能得到恢复甚至提高,或者将CeO2与其它的金属氧化物复合,也能大大提高催化剂的稳定性。王幸宜等[24]采用溶胶-凝胶法制备了不同锰/铈比例的MnOx-CeO2复合金属氧化物,将其用于氯苯(CB)的催化燃烧。研究表明,各种MnOx-CeO2都具有高的催化燃烧氯苯的活性,其中MnOx(0.86)-CeO2活性最高,对1 000×10-6的氯苯完全燃烧的温度仅为254 ℃。氯苯在MnOx-CeO2存在下燃烧的产物为HCl、Cl2、CO2和痕量的CO, 多氯化合物未检测到。较高锰/铈(Mn/Ce+Mn)比的复合金属氧化物有很高稳定活性,这归功于其优异的移除吸附氯物种的能力和大量的活性表面氧。不仅锰/铈配比会影响催化剂的性能,形貌与结构也会影响其催化活性。赵培等[18]考察了CeO2的形貌对MnOx/CeO2催化剂催化燃烧氯苯的影响,结果表明纳米颗粒形态的MnOx/CeO2催化燃烧氯苯的活性远高于纳米棒状的催化剂,原因是纳米颗粒状的催化剂中Mn4+含量更高、氧空位和表面吸附氧更多。 除锰、铈外,其它过渡金属如钴、锆、钒、钛等的氧化物也可用于CVOCs的催化燃烧。在这些金属氧化物中,钒、钛复合金属氧化物(VOx/TiO2)对催化氧化含氯化合物的活性与铬氧化物相当,但稳定性要好得多。TiO2基V2O5-WO3本来是用于选择性催化还原氮氧化物(NOx)的高效催化剂,后用作催化燃烧CVOCs的催化剂,催化活性也较高。Finocchio等[31]研究了高钒含量催化剂对含氯化合物如氯苯、1,2-二氯苯和1,4-二氯苯的催化燃烧,发现V2O5(5%)-WO3-TiO2和V2O5(3.5%)-MoO3-TiO2对完全氧化氯苯有很高的活性。 钙钛矿型复合氧化物(ABO3)是一类具有特定物理和化学性质的新兴催化剂,其结构非常稳定,晶型完整,故氧化还原活性不高。但另一方面,结构中的A和B原子可被半径相近的其它金属离子部分取代而晶体结构保持不变,钙钛矿结构产生缺陷位,原子的氧化态发生变化,从而在低温下即能产生较多的活性氧物种,其氧化还原能力增加,使得具有催化燃烧的活性。 表3 催化燃烧 CVOCs 的典型钙钛矿型复合金属氧化物催化剂 注:VC, vinyl chloride, 氯乙烯。 Poplawski等[37]研究了多种ABO3钙钛矿型氧化物( A=La, Y, Nd 或Gd; B=Fe, Mn, Cr或Co)对1,2-二氯苯(DCB)的催化燃烧活性,发现YCrO3的活性最高,且连续稳定运行几小时后,未见其活性下降。除LaCoO3外,其余的钙钛矿在反应过程中仍保持原来的晶体结构。将AMnO3( A=La, Didym-Di)负载到氧化锆和堇青石载体上[38],发现AMnO3催化氧化含氯化合物的活性,而且副产物的生成量大减少。在ABO3型氧化物中,LaMnO3的热稳定性和氧移动性最高[39],其催化氧化挥发性有机物的活性也最好。但将LaMnO3用于CVOCs的催化氧化时,生成的副产物易使其中毒,因此直接将ABO3型氧化物催化氧化CVOCs的研究不多。Zhang等[34]采用传统的共沉淀法制备了LaMnO3和LaB0.2Mn0.8O3(B=Co, Ni, Fe),将催化燃烧氯乙烯,发现LaB0.2Mn0.8O3的活性要高于纯的LaMnO3钙钛矿型氧化物催化活性的提高源于B元素(Co, Ni, Fe)的低温还原性,以及大量吸附态的氧物种和表面氧空位。 催化燃烧CVOCs的催化剂除了在低温下具有高氧化活性和对CO2、HCl的选择性外,必须有显著的抗失活能力。催化剂的失活路径取决于催化剂的特性(负载贵金属、过渡金属氧化物、钙钛矿氧化物、分子筛等),以及过程操作参数如温度、空速、CVOCs的浓度等。通常情况下,CVOCs的催化氧化失活有以下几种原因:活性相的挥发、催化剂结构改变致中毒、积碳和热分解等[23]。 反应过程中生成的氯与催化剂表面的金属作用会生成金属酰氯,导致活性材料的流失[40]。过渡金属氧化物,特别是铬(Cr)容易因此而失活[41]。用三氯乙烯(TCE)为氯源,50 h后,转化率从77%降至35%,而用氯乙烯时其活性从98%降至93%,这与流失的铬含量一致[42], 铬的流失是由于生成了CrO2Cl2(m.p.-96.5 ℃, b.p.117 ℃)。其它催化剂也会在活性相的损失而失活,如V2O5/SiO2-TiO2在氧化三氯乙烯(TCE)的过程中,钒的含量由反应前的5%降至反应后的1.7%[41],在钙钛矿型催化剂LaCoO3上会沉积CoCl2和Co3O4,将降低其活性相使催化剂失活[43]。对于贵金属催化剂,一般不会因为活性相的挥发而失活。 氯与催化剂之间的相互作用是影响催化剂性能的关键因素,制约催化剂的应用。催化氧化CVOCs活性最高的催化剂是负载在氧化铝上的贵金属,但需要高的反应温度以提高反应速率和消除氯中毒的影响。但是,高温下,生成的HCl会破坏氧化铝载体,导致催化剂失活。将贵金属负载在沸石上,金属和阳离子都对CVOCs的转化起作用,Guillemot等[44]研究了Pt负载在3种沸石上(1.2% Pt/H-Y, 1% Pt/Na-X, 0.65% Pt/Na-Y)催化氧化四氯乙烯(PCE)过程中的失活行为,发现位于III位点上的Na+离子易于和Cl反应,催化剂活性下降,而位于II位点上的Na+离子不易氯化。通常情况下,过渡金属氧化物催化剂的活性要低于贵金属,但不易氯中毒,因此有望取代贵金属成为最有潜力的催化燃烧CVOCs的催化剂。 碳沉积往往发生在氧化挥发性有机物的过程中,这会导致催化剂活性降低[45]。焦碳的形成过程十分复杂,包括催化和非催化反应,一般认为不饱和的含氯化合物容易积碳。在CVOCs的氧化过程中,可能生成不同的不饱和含氯副产物,如氯苯氧化时有多氯苯[23]、三氯乙烯氧化时有四氯乙烯[46]、二氯乙烯氧化时有氯乙烯生成[45]。副产物的分布取决于贵金属的特性和操作工艺参数。Miranda等[46]发现以Ru/Al2O3为催化剂氧化三氯乙烯时,氯代甲烷(PCMs)是主要的氯副产物,表明不饱和化合物容易积碳。酸催化剂如沸石也会受到积碳的影响,酸活性位点能够强烈地吸附反应物,有足够长的时间发生后续结焦反应,结焦反应的速率与酸性位点的数量有关,酸性位点越多,结焦速率越快[47]。结焦多发生在沸石的孔内,通过活性位覆盖和孔堵塞使催化剂活性损失。当催化剂长时间曝露在高温下,会发生热降解而改变催化剂的结构、形貌和理化特性,从而导致催化剂失去原来的活性。 如上所述,导致催化剂失活的原因有几种[48],因此必须考虑如何使催化剂再生,这一课题相对较新,现有的研究报道不多。由于在反应过程中活性相的挥发或者催化剂理化性质的改变(比表面积、结晶度和酸度等)而导致的失活是不可逆的,这类失活无法再生。相对的是,由于氯中毒和结焦导致的失活的催化剂其活性可以部分得到恢复。再生氯中毒的催化剂常用的方法是在高温下通入干空气或湿空气,煅烧一定时间,同时须保证没有含氯化合物存在。Vu等[23]报道Mn-Cu复合金属氧化物在350 ℃下通干空气的氛围中煅烧5 h,其活性几乎可以完全恢复。Dai等[19]认为从催化剂表面完全移除氯物种是缓慢的过程,但如果用湿空气吹扫足够长时间,可以完全移除氯从而使CO2的活性得到恢复。Gallastegi等[49]评估了用氧燃烧处理的方法再生H型沸石催化剂的可行性,发现湿空气比干空气更能有效地促使催化剂活性再生,500 ℃时湿空气氛围中煅烧,既可以除去积碳(氧化或气化),又可以移除氯。经过几次再生循环,H型沸石的催化剂能够完全恢复。 利用非均相催化氧燃烧含氯挥发性有机化合物(CVOCs)是近年内的研究热点,因为其能耗低、效率高,适用范围广。在有催化剂作用下,燃烧CVOCs的温度大致在100~500 ℃, 远低于直接燃烧所需要的温度,且可以完全氧化,氧化产物绝大部分为CO2、H2O和HCl, 极少有Cl2生成,不会导致二次污染。在20世纪90年代和21世纪之初,贵金属和基于贵金属的复合金属氧化物催化剂最先用于CVOCs的催化脱除,铂(Pt)和钯(Pd)具有较高的催化活性,它们一般负载在高比表积的载体如γ-Al2O3和SiO2上,即使很少的负载量(0.1%~0.5%)也能保证CVOCs的燃烧。但是,由于贵金属价贵,且易于氯中毒,使其应用受到极大限制。因此,渡金属如铬(Cr)、锰(Mn)、铁(Fe)、铈(Ce)等价格低廉、环境友好的氧化物成为近年来CVOCs催化脱除的研究热点。由于其比活性相对贵金属要低,因此其负载量稍高(5%~20%), 不容易中毒。氧化铬是已有报道中活性最高的过渡金属氧化物,但在低温下易形成剧毒的残余物,如铬酰氯,故其实用意义不大。 催化剂失活是工业应用的一大障碍,活性位的挥发、Cl2/HCl的强吸附、多孔结构上的积碳等是导致催化剂中毒的几种主要形式。过渡金属氧化物,特别是铬和钒容易因形成金属酰氯而失活。如何克服催化燃烧CVOCs的过程中催化剂和氯作用是一大主要难题。过渡金属氧化物比贵金属催化剂更耐氯中毒,虽然其活性稍低。铈基催化剂催化活性高,但容易吸附HCl/Cl2而失去活性。此外,积碳的生成也是导致催化剂活性下降的重要原因。在未来的研究中,提高催化剂抗氯中毒和耐结焦性能是催化燃烧含氯挥发性有机物领域迫切需要解决的问题,另一方面,结合贵金属、过渡金属氧化物和分子筛等的优点,开发出既具有优异的催化活性、又有良好的稳定性和选择性、不易中毒的新型高效催化剂,将是研究的重点。 参考文献: [1]Yasuhara A, Morita M. Formation of chlorinated compounds in pyrolysis of trichloroethylene[J]. Chemosphere, 1990, 21(4/5): 479-486 [2]Couté N, Richardson J T. Steam reforming of chlorocarbons: Chlorinated aromatics[J]. Applied Catalysis B-Environmental, 2000, 26(3): 217-226 [3]Chen N, Rioux R M, Ribeiro F H. Investigation of reaction steps for the hydrodechlorination of chlorine-containing organic compounds on Pd catalysts [J]. Journal of Catalysis, 2002, 211(1): 192-197 [4]Cardona A I, Candal R, Sánchez B,etal. TiO2on magnesium silicate monolith: Effects of different preparation techniques on the photocatalytic oxidation of chlorinated hydrocarbons[J]. Energy, 2004, 29(5/6): 845-852 [5]Aranzabal A, Pereda-Ayo B, González-Marcos M P,etal. State of the art in catalytic oxidation of chlorinated volatile organic compounds [J]. Chemical Papers, 2014, 68(9): 1 169-1 186 [6]Huang H, Dai Q, Wang X. Morphology effect of Ru/CeO2catalysts for the catalytic combustion of chlorobenzene[J]. Applied Catalysis B-Environmental, 2014, 158/159: 96-105 [7]Matejova L, Topka P, Kaluza L,etal. Total oxidation of dichloromethane and ethanol over ceria-zirconia mixed oxide supported platinum and gold catalysts[J]. Applied Catalysis B-Environmental, 2013, 142: 54-64 [8]Pitkaaho S, Nevanpera T, Matejova L,etal. Oxidation of dichloromethane over Pt, Pd, Rh, and V2O5catalysts supported on Al2O3, Al2O3-TiO2and Al2O3-CeO2[J]. Applied Catalysis B-Environmental, 2013, 138: 33-42 [9]Pitkaaho S, Matejova L, Jiratova K,etal. Oxidation of perchloroethylene-activity and selectivity of Pt, Pd, Rh, and V2O5catalysts supported on Al2O3, Al2O3-TiO2and Al2O3-CeO2[J]. Applied Catalysis B-Environmental, 2012, 126: 215-224 [10]Pitkäaho S, Ojala S, Maunula T,etal. Oxidation of dichloromethane and perchloroethylene as single compounds and in mixtures [J]. Applied Catalysis B-Environmental, 2011, 102(3/4): 395-403 [11]Giraudon J M, Elhachimi A, Leclercq G. Catalytic oxidation of chlorobenzene over Pd/perovskites[J]. Applied Catalysis B-Environmental, 2008, 84(1/2): 251-261 [12]Scire S, Minico S, Crisafulli C. Pt catalysts supported on H-type zeolites for the catalytic combustion of chlorobenzene[J]. Applied Catalysis B-Environmental, 2003, 45(2): 117-125 [13]Gonzalez-Velasco J R, Aranzabal A, Gutierrez-Ortiz J I,etal. Activity and product distribution of alumina supported platinum and palladium catalysts in the gas-phase oxidative decomposition of chlorinated hydrocarbons[J]. Applied Catalysis B-Environmental, 1998, 19(3/4): 189-197 [14]Lopez-Forlseca R, Gutierrez-Ortiz J I, Gonzalez-Velasco J R. Catalytic combustion of chlorinated hydrocarbons over H-BETA and PdO/H-BETA zeolite catalysts [J]. Applied Catalysis A-General, 2004, 271(1/2): 39-46 [15]Wang Y, Jia A, Luo M,etal. Highly active spinel type CoCr2O4catalysts for dichloromethane oxidation [J]. Applied Catalysis B-Environmental, 2015, 165: 477-486 [16]Yang P, Yang S, Shi Z,etal. Deep oxidation of chlorinated VOCs over CeO2-based transition metal mixed oxide catalysts [J]. Applied Catalysis B-Environmental, 2015, 162: 227-235 [17]Yang P, Meng Z, Yang S,etal. Highly active behaviors of CeO2-CrOxmixed oxide catalysts in deep oxidation of 1,2-dichloroethane [J]. Journal of Molecular Catalysis A-Chemical, 2014, 393: 75-83 [18]Zhao P, Wang C, He F,etal. Effect of ceria morphology on the activity of MnOx/CeO2catalysts for the catalytic combustion of chlorobenzene [J]. RSC Advances, 2014, 4(86): 45 665-45 672 [19]Dai Y, Wang X, Dai Q,etal. Effect of Ce and La on the structure and activity of MnOxcatalyst in catalytic combustion of chlorobenzene [J]. Applied Catalysis B-Environmental, 2012, 111: 141-149 [20]de Rivas B, Lopez-Fonseca R, Jimenez-Gonzalez C,etal. Synthesis, characterisation and catalytic performance of nanocrystalline Co3O4for gas-phase chlorinated VOC abatement[J]. Journal of Catalysis, 2011, 281(1): 88-97 [21]Tian W, Fan X, Yang H,etal. Preparation of MnOx/TiO2composites and their properties for catalytic oxidation of chlorobenzene [J]. Journal of Hazardous Materials, 2010, 177(1/3): 887-891 [22]Tseng T K, Wang L, Ho C T,etal. The destruction of dichloroethane over a gamma-alumina supported manganese oxide catalyst [J]. Journal of Hazardous Materials, 2010, 178(1/3): 1 035-1 040 [23]Vu V H, Belkouch J, Ould-Dris A,etal. Removal of hazardous chlorinated VOCs over Mn-Cu mixed oxide based catalyst[J]. Journal of Hazardous Materials, 2009, 169(1/3): 758-765 [24]Wang X, Kang Q, Li D. Catalytic combustion of chlorobenzene over MnOx-CeO2mixed oxide catalysts [J]. Applied Catalysis B-Environmental, 2009, 86(3/4): 166-175 [25]de Rivas B, Lopez-Fonseca R, Sampedro C,etal. Catalytic behaviour of thermally aged Ce/Zr mixed oxides for the purification of chlorinated VOC-containing gas streams [J]. Applied Catalysis B-Environmental, 2009, 90(3/4): 545-555 [26]Dai Q, Wang X, Lu G. Low-Temperature catalytic combustion of trichloroethylene over cerium oxide and catalyst deactivation[J]. Applied Catalysis B-Environmental, 2008, 81(3/4): 192-202 [27]De Rivas B, Lopez-Fonseca R, Gutierrez-Ortiz M A,etal. Catalytic performance of chlorinated Ce/Zr mixed oxides for Cl-VOC oxidation[J]. WIT Transactions on Ecology and the Environment, 109, 857-866. DOI: 10.2495/wm080871 [28]Dai Q, Wang X, Lu G. Low-Temperature catalytic destruction of chlorinated VOCs over cerium oxide [J]. Catalysis Communications, 2007, 8(11): 1 645-1 649 [29]Debecker D P, Bertinchamps F, Blangenois N,etal. On the impact of the choice of model VOC in the evaluation of V-based catalysts for the total oxidation of dioxins: Furanvs. chlorobenzene [J]. Applied Catalysis B-Environmental, 2007, 74(3/4): 223-232 [30]Dobber D, Kiessling D, Schmitz W,etal. MnOx/ZrO2catalysts for the total oxidation of methane and chloromethane [J]. Applied Catalysis B-Environmental, 2004, 52(2): 135-143 [31]Finocchio E, Ramis G, Busca G. A study on catalytic combustion of chlorobenzenes [J]. Catalysis Today, 2011, 169(1): 3-9 [32]Lu Y, Dai Q, Wang X. Catalytic combustion of chlorobenzene on modified LaMnO3catalysts[J]. Catalysis Communications, 2014, 54: 114-117 [33]Chen S, Wang Y, Jia A,etal. Enhanced activity for catalytic oxidation of 1,2-dichloroethane over Al-substituted LaMnO3perovskite catalysts [J]. Applied Surface Science, 2014, 307: 178-188 [34]Zhang C, Wang C, Zhan W,etal. Catalytic oxidation of vinyl chloride emission over LaMnO3and LaB0.2Mn0.8O3(B=Co, Ni, Fe) catalysts [J]. Applied Catalysis B-Environmental, 2013, 129: 509-516 [35]沈柳倩, 翁芳蕾, 袁鹏军, 等. 钙钛矿型催化剂对VOCs催化燃烧的抗毒性和稳定性研究 [J]. 分子催化, 2008, 22(4): 320-324 Shen Liuqian, Weng Fanglei, Yuan Pengjun,etal. Research on the poison resistance and stabilization of the perovskite catalysts for VOCs catalytic combustion [J]. Journal of Molecular Catalysis (China), 2008, 22(4): 320-324(in Chinese) [36]Sinquin G, Petit C, Libs S,etal. Catalytic destruction of chlorinated C-2 compounds on a LaMnO3+delta perovskite catalyst [J]. Applied Catalysis B-Environmental, 2001, 32(1/2): 37-47 [37]Poplawski K, Lichtenberger J, Keil F J,etal. Catalytic oxidation of 1,2-dichlorobenzene over ABO3-type perovskites [J]. Catalysis Today, 2000, 62(4): 329-336 [38]Stephan K, Hackenberger M, Kiessling D,etal. Supported perovskite-type oxide catalysts for the total oxidation of chlorinated hydrocarbons[J]. Catalysis Today, 1999, 54(1): 23-30 [39]Najjar H, Batis H. La-Mn perovskite-type oxide prepared by combustion method: Catalytic activity in ethanol oxidation [J]. Applied Catalysis A-General, 2010, 383(1/2): 192-201 [40]Abdullah A Z, Abu Bakar M Z, Bhatia S. Combustion of chlorinated volatile organic compounds (VOCs) using bimetallic chromium-copper supported on modified H-ZSM-5 catalyst [J]. Journal of Hazardous Materials, 2006, 129(1/3): 39-49 [41]Kulazynski M, van Ommen J G, Trawczynski J,etal. Catalytic combustion of trichloroethylene over TiO2-SiO2supported catalysts [J]. Applied Catalysis B-Environmental, 2002, 36(3): 239-247 [42]Rachapudi R, Chintawar P S, Greene H L. Aging and structure activity characteristics of CR-ZSM-5 catalysts during exposure to chlorinated VOCs [J]. Journal of Catalysis, 1999, 185(1): 58-72 [43]Kiessling D, Schneider R, Kraak P,etal. Perovskite-type oxides catalysts for the total oxidation of chlorinated hydrocarbons [J]. Applied Catalysis B-Environmental, 1998, 19(2): 143-151 [44]Guillemot M, Mijoin J, Mignard S,etal. Mode of zeolite catalysts deactivation during chlorinated VOCs oxidation [J]. Applied Catalysis A-General, 2007, 327(2): 211-217 [45]Aranzabal A, Gonzalez-Marcos J A, Romero-Saez A,etal. Stability of protonic zeolites in the catalytic oxidation of chlorinated VOCs (1,2-dichloroethane) [J]. Applied Catalysis B-Environmental, 2009, 88(3/4): 533-541 [46]Miranda B, Diaz E, Ordonez S,etal. Performance of alumina-supported noble metal catalysts for the combustion of trichloroethene at dry and wet conditions [J]. Applied Catalysis B-Environmental, 2006, 64(3/4): 262-271 [47]Guisnet M, Magnoux P. Fundamental description of deactivation and regeneration of acid zeolites[C]//Delmon B, Froment G F: Catalyst Deactivation, 1994, 88: 53-68 [48]Aranzabal A, Romero-Saez M, Elizundia U,etal. Deactivation of H-zeolites during catalytic oxidation of trichloroethylene [J]. Journal of Catalysis, 2012, 296: 165-174 [49]Gallastegi-Villa M, Romero-Saez M, Aranzabal A,etal. Strategies to enhance the stability of H-beat zeolite in the catalytic oxidation of Cl-VOCs: 1,2-Dichloroethane [J]. Catalysis Today, 2013, 213: 192-1971.2 过渡金属氧化物
1.3 钙钛矿型复合氧化物催化剂
2 催化剂的失活与再生
2.1 催化剂的失活
2.2 催化剂再生
3 结论与展望
猜你喜欢
中学生数理化·自主招生(2022年4期)2022-05-09 22:00:23
中国果业信息(2021年5期)2021-12-05 22:10:28
环境卫生工程(2021年1期)2021-03-19 05:22:42
恋爱婚姻家庭·养生版(2020年11期)2020-12-17 03:26:48
中国盐业(2018年16期)2018-12-23 02:08:28
劳动保护(2018年8期)2018-09-12 01:16:20
合成化学(2015年10期)2016-01-17 08:56:24
浙江科技学院学报(2014年6期)2014-02-28 22:12:09
中国氯碱(2014年10期)2014-02-28 01:04:59
河南科技(2014年8期)2014-02-27 14:07:42
