
碳酸钙热分解进展
2015-04-01 11:53:54卢尚青吴素芳
化工学报 2015年8期
卢尚青,吴素芳
(浙江大学化学工程与生物工程学院,浙江 杭州 310027)
引言
由于化石燃料的大量使用,产生CO2等温室气体随意排放造成的全球变暖,严重地影响到了地球的生态环境和全球气候[1-2]。因此,研究CO2减排在全球各国具有紧迫而重要的意义。
利用 CaO反应吸附脱除CO2,以及 CaCO3分解为CaO与CO2的钙循环过程,是工业干法脱碳的重要手段。该技术不仅可用于烟气脱碳,如燃烧后钙基循环脱碳[3-5],还可用于化学化工过程,如甲烷蒸气反应吸附强化制氢[6-7]。利用CaO作为高温CO2吸附剂是因为CaO具有较高的CO2摩尔吸附容量;其次,CaO可从自然界中广泛存在的天然石灰石中获得,其脱碳成本仅为甲醇胺(MEA)溶液吸附脱碳的一半[8],具有极大的经济优势。CaO 基CO2吸附剂的成本又是影响钙循环脱碳成本的重要因素。高温CaO基CO2吸附剂重复循环使用,是降低钙循环脱碳技术成本的关键。因此,需要对CaO基CO2吸附剂重复使用过程中 CaCO3热分解再生性能和影响因素进行研究。
目前,国内外对CaO基吸附剂的研究主要集中在CaO反应吸附CO2,即碳酸化过程[4-5],而对CaO循环利用的另一半,即CaCO3的热分解[9-12],研究缺乏系统性。而目前普通的CaCO3需要900℃以上的高温热分解再生,导致过程能耗高,严重限制其优势的发挥。因此,本文首次对CaCO3热分解平衡及其机理,CaCO3材料的粒径、显微结构、添加物,以及热分解操作条件等几方面,综述CaCO3热分解反应过程的分解温度和分解速率特性,特别是对微米及纳米CaCO3的热分解反应进行分析综述,为今后研究提供参考。
1 碳酸钙热分解机理及分解平衡
CaCO3的热分解属于典型的固态分解反应,且反应可逆,其反应式为

研究者普遍认为其分解过程包括以下5个步骤:
(1)环境中的热量传递到颗粒的表面;
(2)颗粒表面的热量传递到颗粒的反应内表面;
(3)在反应内表面上进行热吸收和热分解;
(4)分解形成的CO2在无孔CaO层和CaO层内孔扩散;
(5)CO2从颗粒的内表面扩散到环境中。
其中步骤(1)和(2)的热量传递影响着CaCO3的分解;步骤(4)和(5),传质则是其主要影响因素;而 CaCO3的化学反应则由步骤(3)决定。因此,CaCO3的热分解反应受传热、传质以及化学反应3个因素的影响,而这3个影响因素又与CaCO3的颗粒粒径、显微结构等有重要关系。此外,加热速率、分解气氛和分解压力等外部操作条件亦会对分解产生影响。
对CaCO3热分解过程进行反应平衡研究,根据经典物理化学,在一定温度下,CaCO3发生分解首先需满足其 CO2平衡分压大于分解气氛中的 CO2分压,否则,CaCO3的分解反应将不会发生[13]。CaCO3分解产生 CO2的平衡分压与温度的关系如图 1[13]所示,图中的点为实验测得的平衡分压,直线为拟合值。在能发生热分解反应的条件下,根据固态化合物分解反应特性,可以认为反应最初发生在某些区域的一点上,随着反应进行,这些邻近的星星点点的分解产物逐渐聚积成一个个新物相的核,被称为分解晶核,随后核周围的分子继续在核上发生界面反应,持续进行,直到整个固相完全分解[14]。
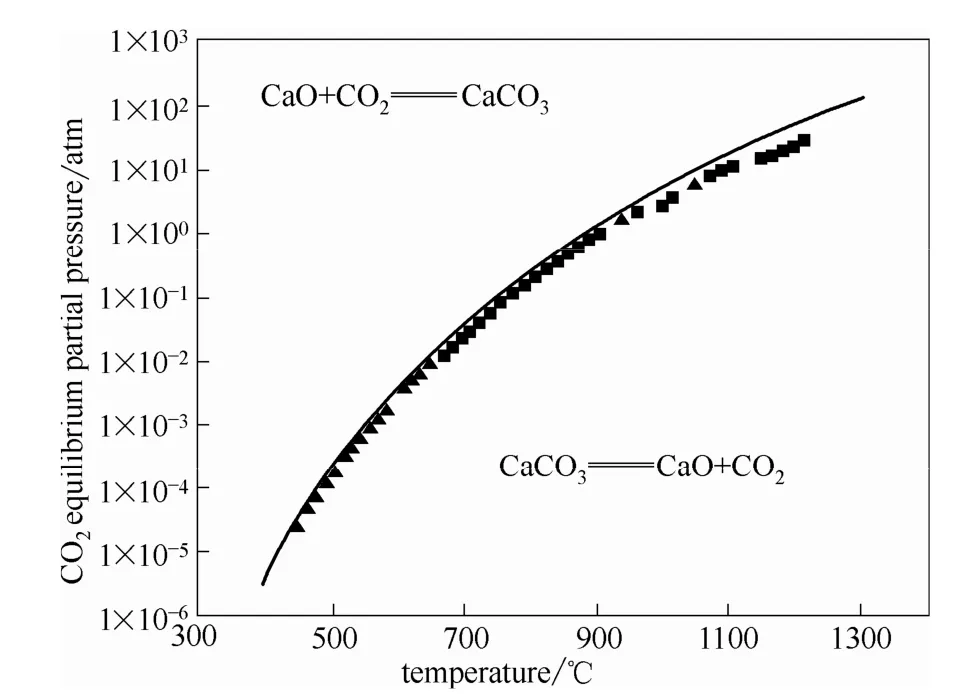
图1 CO2平衡分压与CaCO3分解温度的关系Fig.1 Relationship between CO2equilibrium partial pressure and CaCO3decomposition temperature (400—1300℃)
2 粒径对碳酸钙热分解反应的影响
粒径对 CaCO3热分解产生影响主要是因为其与分解过程中的传热、传质及化学反应均有密切关系。以传质为例,CaCO3热分解过程中最主要的传质阻力就是CO2通过无孔CaO层及CaO层内孔的扩散。随着CaCO3颗粒粒径的减小,CO2通过CaO层的内扩散阻力亦随之迅速减小。
来蔚鹏等[15]对 CaCO3分解反应热力学性质进行研究,认为随着粒径的减小,CaCO3分解反应的标准摩尔反应焓 ΔrH⊖、标准摩尔反应熵 ΔrS⊖和标准摩尔反应吉布斯函数ΔrG⊖均减小,标准平衡常数K⊖增大,从而使其分解温度降低。
此外,Cui等[16]利用 KAS方法研究了 CaCO3颗粒粒径对其分解活化能的影响,得到结果如图 2所示。可以看出,随着粒径的减小,CaCO3的平均分解活化能呈现下降的趋势。
Wang等[17]在热力学性质的基础上对纳米材料的热分解温度进行了深入的理论研究,并提出一个颗粒半径与分解温度间的简化模型

当颗粒半径大于10 nm,半径变化对表面张力的影响可忽略,式(2)右侧小于0,因此从理论上证明了随着颗粒半径的减小,纳米材料的分解温度降低。此外,其对纳米CaCO3分解温度的实验研究,亦证实分解温度与颗粒半径倒数之间存在线性关系,如图 3[17]所示,进一步证实了该模型的正确性。图中Tonset为起始分解温度,Tmax为最快分解速率对应的分解温度。
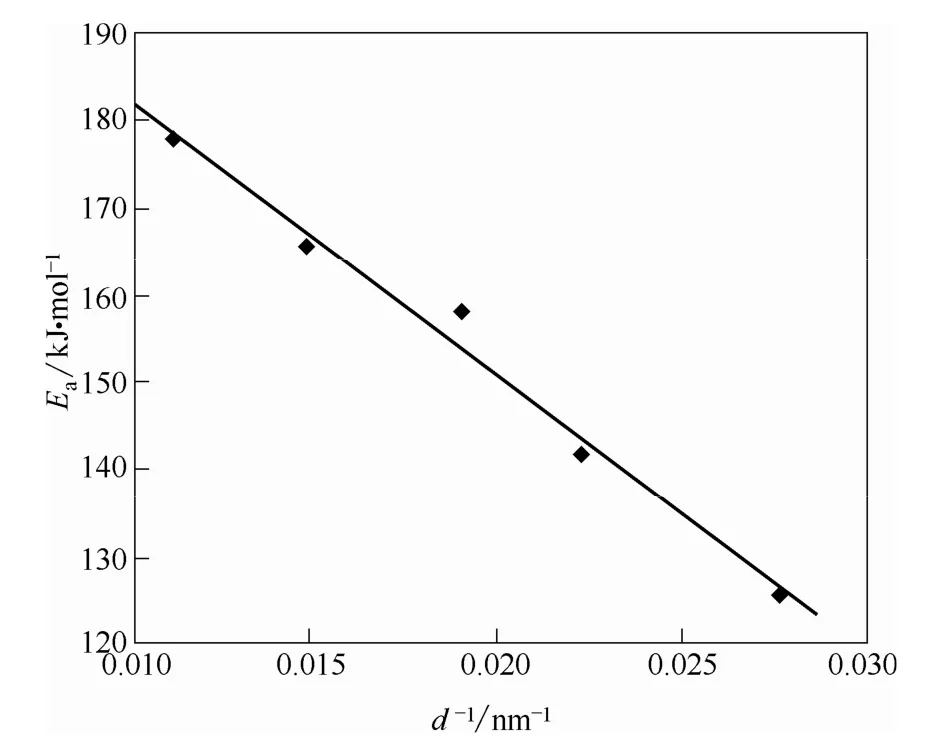
图2 平均活化能与CaCO3颗粒粒径倒数间的关系Fig.2 Relationship between average activation energy and reciprocal of CaCO3particle diameter

图3 分解温度与纳米CaCO3颗粒半径倒数间的关系Fig.3 Relationship between decomposition temperature and reciprocal of particle radii of nano-CaCO3
岳林海等[18-19]研究了粒径为40~80 nm的纳米级CaCO3与粒径为5~20 μm的参比样品的分解特性,发现纳米 CaCO3的起始分解温度降低了约 40℃,最终分解温度降低了约60℃,分解活化能亦有60~80 kJ·mol-1的下降。扫描电镜或透射电镜研究结果发现,纳米微晶之间的团聚使得粒径增大,表面自由能下降,桥键等作用力增强,使分解活化能有所增加,从而证明了粒径减少,分解温度降低的事实。X射线衍射的研究结果发现,超细CaCO3的晶格中存在着较大的晶格畸变应力,晶格不完整程度增加。因此,作者提出晶格缺陷可能导致纳米CaCO3分解活化能下降,从而解释了导致分解温度显著下降的主要原因。
Wu等[20]采用热重分析仪测定并比较70 nm和5 μm的CaCO3的分解温度,发现70 nm纳米CaCO3起始和最终分解温度均比5 μm微米CaCO3下降大约50℃。Liu等[21]亦发现20 nm的CaCO3比分析纯CaCO3(20 μm左右)的分解温度低180℃。以上结果分析认为纳米CaCO3由于粒径小,不仅热量更容易从外界传递进CaCO3内部,分解产生的CO2更容易扩散出去,而且,由于分解速率受界面化学反应控制,纳米CaCO3增大了比表面积,从而提供了更大的分解反应面积,提高了分解反应速率。因此,粒径越小,CaCO3分解温度越低。
综上所述,随着CaCO3颗粒粒径的减小,CO2的内扩散阻力降低,且分解活化能亦降低,使小粒径CaCO3的分解反应更容易发生,因此其分解温度亦愈低,分解速率愈快[22-24],而且随着CaCO3颗粒粒径的减小,化学反应将逐渐成为分解过程的控速步骤。
3 显微结构对碳酸钙分解的影响
颗粒显微结构指颗粒内部孔隙率、孔径以及比表面积等的总体表现。仲兆平等[25]发现粒径 53 μm~1 mm 的石灰石颗粒的煅烧反应速度基本相同,认为CaCO3颗粒在热分解过程中发生爆裂,从而产生出更多的孔结构,使其比表面积增加。此外,Yan等[26]对90~180 μm CaCO3颗粒在升温速率为10℃·min-1时的热分解进行研究,发现 106~150 μm 的煅烧速率反而最快。研究者认为热传递、物质传递和颗粒显微结构之间存在复杂的非线性关系,除了颗粒粒径外,CaCO3颗粒本身的显微结构对其热分解存在重要影响。
Campbell等[27]对受传质、传热控制的大粒径CaCO3颗粒的扩散过程进行研究,认为CaCO3颗粒的孔隙率等显微结构对热分解过程中的CO2扩散速率有影响。根据扩散特点,他们认为此时CO2的扩散属于Knudsen扩散,其扩散系数为
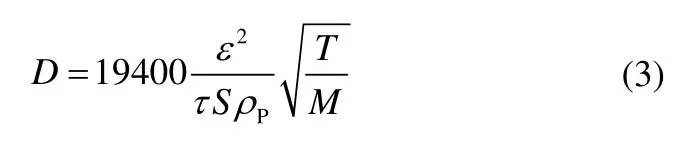
作者据此进一步推导出CaCO3热分解传质模型,结果能很好地预测孔隙率高达0.66时的CaCO3分解特点,结果如图4所示。
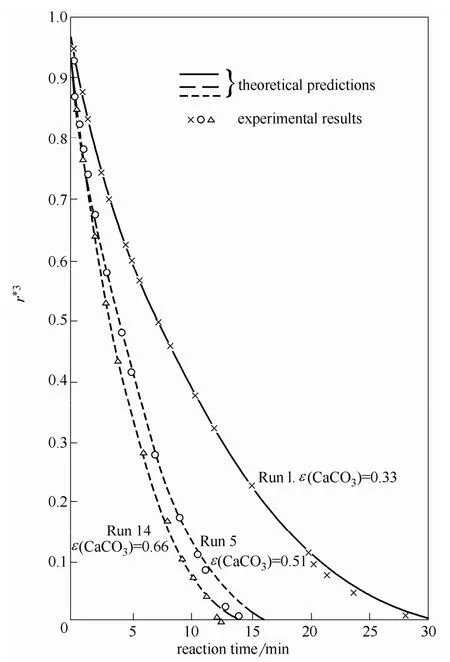
图4 不同孔隙率CaCO3分解速率与反应时间的关系Fig.4 Decomposition rates of CaCO3of varying porosity-fractional unreacted mass against reaction time
Borgwardt等[23]对粒径为 1~90 μm 的 CaCO3的热分解速率进行研究,认为此时CaCO3的热分解受化学反应控制,并指出其分解速率与CaCO3的比表面积密切相关。根据研究,他提出一个热分解初期的反应模型

作者认为CaCO3热分解反应发生在CaO和CaCO3内界面上,而内界面的大小与反应速率相关。对于分解初期,BET测得的比表面积即为反应界面面积,因此颗粒的比表面积对热分解速率有直接影响。
综上所述,CaCO3颗粒的孔隙率、比表面积等显微结构对热分解速率有一定影响。在一定程度上,对受传质过程控制的热分解,大的孔隙率和比表面积有利于促进CaCO3的分解。
4 添加物对碳酸钙分解的影响
对于CaCO3中存在孔隙对热分解的作用,除了上述介绍的对CO2扩散的有利影响外,Huang等[28-29]认为 CaCO3颗粒中存在的间隙阻碍了热量从颗粒的外表面向反应界面的传递。为此,他们提出向CaCO3颗粒中掺杂低熔点物质的方法以减少孔隙。在CaCO3分解温度范围内,Li2CO3、V2O5等物质融熔,颗粒缩小,填充到孔道间隙中,加快热量传递。但是需要注意的是,如果添加物熔点过高,则会阻碍 CaCO3的分解;熔点过低又会堵塞孔道,阻碍CO2的扩散,减缓分解速率。添加剂的部分研究结果见表1。

表1 添加改性CaCO3的分解速率Table 1 Decomposition rate of doped CaCO3(DGA-TGA, 700℃)
此外,余兆南等[30]研究了 K、Na化合物对CaCO3热分解的影响,发现其能提高CaCO3热分解速率和降低分解温度。他们认为这些化合物能与CaO形成低熔点共融物,使 CaO产物层的空隙增加,进而减小了CO2的扩散阻力,促进CaCO3的热分解。
除了添加物质以减少孔隙的研究之外,还有其他的一些添加物对CaCO3分解的研究。侯贵华等[31]研究了CuO对CaCO3分解温度的影响,发现用机械混合法添加1%的CuO能使CaCO3的起始和最终分解温度分别降低12和15℃。他们认为CuO能与CaCO3分解产物反应生成CaCu2O3和Ca2CuO3,使产物层局部或整体的厚度减薄,从而降低热分解中传质阻力,促进其分解。Braileanu等[32]对 CaCO3和Bi2O3混合粉末进行研究,发现少量Bi2O3的存在使得CaCO3的最快分解速率对应的温度由840℃降为790℃。他们认为Bi2O3能与CaO反应形成 Ca6Bi7O16.5,从而改变CO2在产物层的扩散阻力,促进 CaCO3分解。此外,Calvo等[33]研究方解石的纯度对其分解动力学参数的影响,发现杂质的存在虽然不影响模型的选择但能降低热分解活化能。
张惠珠等[34]研究了 AlOOH膜包覆改性后CaCO3的分解性能,发现包覆后CaCO3的分解反应延迟,分解温度亦升高,他们认为 AlOOH包覆层的存在不仅阻碍了外界热量向反应界面的传递,而且也影响分解产物CO2的扩散。
师琦等[35]用沉淀法制备了 SiO2包覆改性的纳米CaCO3,不仅发现其稳定性有所提高,而且分解温度也有所降低,其中,当Si质量分数为4.93%时,纳米CaCO3的最终分解温度比未包覆的降低42℃。他们认为Si能以化学键形式结合在CaCO3表面,形成Si—O—Ca键[36],减弱对CO2的束缚,从而促进CaCO3分解。
王成毓[37]对纳米CaCO3的仿生合成进行研究,发现,在十八醇磷酸酯的存在下,通过碳化法制备得到的纳米 CaCO3的最终分解温度与未加十八醇磷酸酯的样品相比,降低了约30℃。对此,他提出了有机质与CaCO3相互作用机理,认为磷酸酯附着在CaCO3表面,削弱了CaO与CO2的结合键能,使得CaCO3分解温度降低。
综上所述,添加剂的存在对降低CaCO3的分解温度有不同的影响,其原理大致有与CaO共熔减少孔隙,改善传质传热阻力,或与CaO结合削弱键能等几种。
5 操作条件对碳酸钙分解的影响
除了 CaCO3颗粒粒径、显微结构等内部因素外,操作条件对CaCO3的分解影响也是一个重要方面。由于大部分分解都是在常压下进行,因此操作条件主要指加热速率、分解气氛等。
Sanders等[38]对同等粒径CaCO3变温分解过程进行研究,发现不同加热速率下得到的CaCO3分解温度和分解速率均不同,其中当加热速率由 2℃·min-1升至32℃·min-1时,CaCO3的分解温度由677℃变为875℃。Liu等[21]对CaCO3变温分解过程动力学进行研究,发现加热速率为 10℃·min-1时其分解活化能最大。此外,Yan等[26]发现快速煅烧不利于产物 CaO碳酸化速率的提高,反而会导致石灰石钙利用率的降低。但是以上研究均未明确指出加热速率对分解速率的影响。在此基础上,Galan等[39]较为深入研究了加热速率对等粒径CaCO3分解行为的影响,发现 CaCO3的分解温度随着加热速率的变慢而降低,但分解速率加快。作者认为较快的加热速率会使 CaCO3热分解的温度范围变宽,直接导致其分解温度升高,但较快的加热速率意味着较高的温度,使 CaCO3同一时刻的分解速率加快。
对分解气氛的研究中,主要采用的是CO2和水蒸气两种分解气氛。一方面,根据图1结果所示,CO2分压对CaCO3热分解有较大影响,国内外诸多学者[39-42]对其进行相应研究。一般而言,随着分解气氛中CO2分压的增大,CaCO3开始分解时间推迟,分解速率减慢,分解温度增加[43]。另一方面,一些研究表明水蒸气的存在能促进 CaCO3的热分解。Wang等[44]对此进行研究,认为水蒸气能够结合于CaCO3表面,削弱CaO和CO2间的结合键能,从而促进其分解速率提高并降低其分解温度。L’vov等[45]对碱土金属碳酸盐热分解机理的研究亦表明水蒸气能降低此类碳酸盐的分解温度。
总而言之,适当的加热速率、较低的CO2分压、适量的水蒸气存在等有利于CaCO3的热分解。
6 碳酸钙分解反应动力学
CaCO3分解动力学研究的关键是建立合理的分解模型以加深对分解过程的认识。诸多研究者对此过程进行研究,其中 Broda等[46]、Stanmore等[47]及冯云等[48]较为全面地综述了 CaCO3分解动力学模型。根据综述,CaCO3的热分解模型主要有收缩核模型、结构空隙模型、均匀反应模型、修正的收缩核模型、微粒模型及随机孔模型等几种。这些模型均从不同角度阐释了 CaCO3分解动力学的某些特征。一般来说,微粒模型和结构空隙模型被用来描述颗粒内部分解的过程;均匀反应模型适合描述小粒径CaCO3热分解行为;收缩核模型适合模拟致密固相颗粒反应,在忽略烧结的条件下,该模型能较好描述CaCO3的热分解过程。
根据这些模型,不同研究者针对特定的CaCO3分解过程建立了不同的动力学方程,部分结果见表2。
以上分析的 CaCO3分解动力学模型用于微米或纳米CaCO3热分解研究,并形成了较为成熟的理论体系,其中收缩核模型效果最好。然而,目前对纳米CaCO3分解动力学的研究很少,师琦等[56]对纳米 CaO基吸附剂的分解动力学进行了较为深入的研究,采用收缩核模型成功模拟了吸附剂在 600~800℃、N2及含 CO2气氛下的分解行为,得到 N2下的活化能为141.9 kJ·mol-1,与Dennis等[57]计算得到的微米级 CaCO3分解活化能相比,降低了约36 kJ·mol-1。因此,作者认为纳米CaCO3比微米CaCO3容易分解,并体现为分解温度的降低和分解速度加快。

表2 CaCO3分解动力学方程Table 2 Decomposition kinetic equations of CaCO3
7 结 论
CaCO3的热分解属于典型的固态分解反应,其分解平衡受 CO2分压的制约。颗粒粒径对 CaCO3热分解有着决定性影响。随着CaCO3粒径的减小,比表面积增大,CaCO3热分解速率加快,分解温度降低。CaCO3颗粒的显微结构在一定程度上会改变传质传热阻力及反应界面大小等,进而对分解速率和分解温度产生影响。较大的孔隙率能促进受传质控制的CaCO3的热分解,而较大的比表面积能改善受化学反应控制的CaCO3的热分解性能。Li2CO3、V2O5等物质能熔融改善传质,K、Na化合物则能与CaO共融以改善CO2的扩散性能,而CuO、Bi2O3等则可以通过与CaO的反应改善传质性能,上述几种均能促进分解反应的进行,提高分解速率,降低分解温度。添加水蒸气、降低CO2分压等外部操作条件的改变亦促进CaCO3的分解。此外,分解动力学的研究表明,纳米级CaCO3分解速度快于微米级CaCO3的分解速度,收缩核模型能较好模拟其分解特性。
总之,通过本文综述认为,纳米CaCO3具有较低的分解温度,将其作为钙循环吸附剂的前驱体将极大降低再生环节所需温度,节约捕集CO2所需成本,是现在一个重要的研究发展方向。在此基础上,可通过改变吸附剂的显微结构或其他添加物进一步降低钙循环的再生能耗这一瓶颈问题。
符号说明
c——CO2浓度,mol·m-3
D——CO2扩散系数,m2·s-1
d——CaCO3颗粒直径,m
k——CaCO3分解速率常数
M——摩尔质量,g·mol-1
n——物质的量,mol
p——分解压力,Pa
R——CaCO3颗粒半径,m
r——CaCO3的分解速率,mol·m-2·s-1
S——比表面积,m2·g-1
T——温度,K
t——CaCO3分解时间,s
Vm——摩尔体积,L·mol-1
X——CaCO3分解转化率
ε——CaCO3的孔隙率
ν——化学计量数
ρ——密度,kg·m-3
σ——表面张力,N·m-2
τ——黏度系数
[1] Rogelj J, Meinshausen M, Knutti R. Global warming under old and new scenarios using IPCC climate sensitivity range estimates [J].Nature Climate Change, 2012, 2(4):248-253.
[2] Intergovernmental Panel on Climate Change. Climate change 2014:Mitigation of Climate Change[R]. New York:IPCC, 2014.
[3] Mofarahi M, Roohi P, Farshadpoor F. Study of CaO sorbent for CO2capture from flue gases//9th International Conference on Chemical and Process Engineering. Chemical Engineering Transactions[C].Bushehr:Persian Gulf University, 2009:403-408.
[4] Wu S F, Zhu Y Q. Behavior of CaTiO3/nano-CaO as a CO2reactive adsorbent [J].Ind. Eng. Chem. Res., 2010, 49(6):2701-2706.
[5] Rodríguez N, Alonso M, Abanades J C. Average activity of CaO particles in a calcium looping system [J].Chem. Eng. J., 2010, 156(2):388-394.
[6] Han C, Harrison D P. Simultaneous shift reaction and carbon dioxide separation for the direct production of hydrogen [J].Chem. Eng. Sci.,1994, 49(24):5875-5883.
[7] Wu Sufang, Li Lianbao, Zhu Yanqing, Wang Xieqing. A micro-sphere catalyst complex with nano CaCO3precursor for hydrogen production used in ReSER process [J].Engineering Sciences, 2010,8(1):22-26.
[8] Abanades J C, Rubin E S, Anthony E J. Sorbent cost and performance in CO2capture systems [J].Ind. Eng. Chem. Res., 2004, 43(13):3462-3466.
[9] Lu D Y, Hughes R W, Anthony E J, Manovic V. Sintering and reactivity of caco3-based sorbents forin situCO2capture in fluidized beds under realistic calcination conditions [J].J. Environ. Eng., 2009,135(6):404-410.
[10] Manovic V, Charland J, Blamey J, Fennell P S, Lu D Y, Anthony E J.Influence of calcination conditions on carrying capacity of CaO-based sorbent in CO2looping cycles [J].Fuel, 2009, 88(10):1893-1900.
[11] Alvarez D, Pena M, Borrego A G. Behavior of different calcium-based sorbents in a calcination/carbonation cycle for CO2capture [J].Energy Fuels, 2007, 21(3):1534-1542.
[12] Lu H, Reddy E P, Smirniotis P G. Calcium oxide based sorbents for capture of carbon dioxide at high temperatures [J].Ind. Eng. Chem.Res., 2006, 45(11):3944-3949.
[13] Florin N H, Harris A T. Enhanced hydrogen production from biomass within situcarbon dioxide capture using calcium oxide sorbents [J].Chem. Eng. Sci., 2008, 63(2):287-316.
[14] Su Mianzeng(苏勉曾).Solid Chemistry Introduction(固体化学导论)[M]. Beijing:Beijing University Press, 1986.
[15] Lai Weipeng (来蔚鹏), Xue Yongqiang (薛永强), Lian Peng (廉鹏),Ge Zhongxue (葛忠学), Wang Bozhou (王伯周), Zhang Zhizhong(张志忠). Effect of particle size on properties of chemical reaction thermodynamics of nanosystems [J].Journal of Physical Chemistry(物理化学学报), 2007, 23(4):508-512.
[16] Cui Z, Xue Y, Xiao L, Wang Tingting. Effect of particle size on activation energy for thermal decomposition of nano-CaCO3[J].J.Comput. Theor. Nanos., 2013, 10(3):569-572.
[17] Wang S, Cui Z, Xia X, Xue Y. Size-dependent decomposition temperature of nanoparticles:a theoretical and experimental study [J].Phys. B:Condens. Matter., 2014, 454:175-178.
[18] Yue Linhai (岳林海), Shui Miao (水淼), Xu Zhude(徐铸德).Decomposition kinetics of nano-particle calcite [J].Chinese Journal of Inorganic Chemistry(无机化学学报), 1999, 15(2):225-228.
[19] Yue Linhai (岳林海), Shui Miao (水淼), Xu Zhude(徐铸德). The crystal structure of ultra-fine CaCO3and its thermal decomposition[J].Journal of Chemical Engineering of Chinese Universities(高校化学工程学报),2000, 21(10):1555-1559.
[20] Wu S F, Li Q H, Kim J N, Yi, Kwang B. Properties of a nano CaO/Al2O3CO2sorbent[J].Ind. Eng. Chem. Res., 2008, 47(1):180-184.
[21] Liu R, Chen J, Guo F, Yun Jimmy, Shen Z. Kinetics and mechanism of decomposition of nano-sized calcium carbonate under non-isothermal condition [J].Chin. J. Chem. Eng., 2003, 11(3):302-306.
[22] Salvador A R, Calvo E G, Aparicio C B. Effects of sample weight,particle size, purge gas and crystalline structure on the observed kinetic parameters of calcium carbonate decomposition [J].Thermochim. Acta., 1989, 143:339-345.
[23] Borgwardt R H. Calcination kinetics and surface area of dispersed limestone particles [J].AIChE J., 1985, 31(1):103-111.
[24] Criado J M, Ortega A. A study of the influence of particle size on the thermal decomposition of CaCO3by means of constant rate thermal analysis [J].Thermochim. Acta., 1992, 195:163-167.
[25] Zhong Zhaoping (仲兆平), Marnie Telfer, Zhang Mingyao (章名耀),Li Daji (李大骥), Xu Yuenian (徐跃年), Jin Baosheng (金保升), Lan Jixiang (兰计香), Zhang Dongke (张东柯). Experimental study on pyrolysis of caroline linestone [J].Journal of Combustion Science and Technology(燃烧科学与技术),2001, 7(2):110-114.
[26] Yan C, Grace J R, Lim C J. Effects of rapid calcination on properties of calcium-based sorbents [J].Fuel Process. Technol., 2010, 91(11):1678-1686.
[27] Campbell F R, Hills A, Paulin A. Transport properties of porous lime and their influence on the decomposition of porous compacts of calcium carbonate [J].Chem. Eng. Sci., 1970, 25(6):929-942.
[28] Huang J, Daugherty K E. Lithium carbonate enhancement of the calcination of calcium carbonate:proposed extended-shell model [J].Thermochim. Acta., 1987, 118:135-141.
[29] Huang J, Daugherty K E. Inhibition of the calcination of calcium carbonate [J].Thermochim. Acta., 1988, 130:173-176.
[30] Yu Zhaonan(余兆南). The experimental study of CaCO3decomposition [J].Journal of Thermal Energy and Power Engineering(热能动力工程), 1997, 12(4):278-280.
[31] Hou Guihua (侯贵华), Shen Xiaodong (沈晓冬), Xu Zhongzi (许仲梓). Effect of copper oxide on decomposition kinetics for calcium carbonate [J].Journal of the Chinese Ceramic Society(硅酸盐学报),2005, 33(1):109-114.
[32] Braileanu A, Zaharescu M, CriŞan D, FĂtu D, Segal E, Danciulescu C. Kinetics of the decomposition of calcium carbonate in the presence of Bi2O3[J].J. Therm. Anal., 1996, 47(2):569-575.
[33] Calvo E G, Arranz M A, Leton P. Effects of impurities in the kinetics of calcite decomposition [J].Thermochim. Acta., 1990, 170:7-11.
[34] Zhang Huizhu (张惠珠), Jin Dalai (金达莱), Yue Linhai (岳林海).Study on the non-isothermal decomposition kinetics of AlOOH coated calcium carbonate [J].Journal of Zhejiang University:Science Edition(浙江大学学报:理学版),2008, (4):16.
[35] Shi Qi (师琦), Wu Sufang (吴素芳). Properties of SiO2coated nano SiO2/CaCO3sorbents by precipitation method [J].CIESC Journal(化工学报), 2009, 60(2):507-513.
[36] Yue Linhai (岳林海), Cai Juxiang (蔡菊香), Hua Yimiao (华益苗).Mechanism and structure of SiO2-coated CaCO3superfine particles[J].Journal of Zhejiang University:Science Edition(浙江大学学报:理学版), 2002, 29(1):67-72.
[37] Wang Chengyu (王成毓). Biomimetic synthesis and character of functional nano-CaCO3[D]. Changchun:Jilin University, 2007.
[38] Sanders J P, Gallagher P K. Kinetic analyses using simultaneous TG/DSC measurements (Ⅰ):Decomposition of calcium carbonate in argon [J].Thermochim Acta., 2002, 388(1/2):115-128.
[39] Galan I, Glasser F P, Andrade C. Calcium carbonate decomposition[J].J. Therm. Anal Calorim., 2013, 111(2):1197-1202.
[40] Avila I, Crnkovic P M, Milioli F E, Luo Kai H. Thermal decomposition kinetics of Brazilian limestones:effect of CO2partial pressure [J].Environ. Technol.. 2012, 33(10):1175-1182.
[41] Yin J, Kang X, Qin C, Feng B, Veeraragavan A, Saulov D,et al.Modeling of CaCO3decomposition under CO2/H2O atmosphere in calcium looping processes [J].Fuel Process. Technol., 2014, 125:125-138.
[42] L'vov B V. Kinetic parameters of CaCO3decomposition in vacuum,air and CO2calculated theoretically by means of the thermochemical approach[J].React. Kinet. Mech. Cat., 2015, 114(1):31-40.
[43] Li Zhenshan (李振山), Fang Fan (房凡), Cai Ningsheng (蔡宁生).Simulation of CaCO3calcination under high CO2concentration [J].Journal of Engineering for Thermal Energy and Power(热能动力工程), 2007, (6):642-646.
[44] Wang Y, Thomson W J. The effects of steam and carbon dioxide on calcite decomposition using dynamic X-ray diffraction [J].Chem. Eng.Sci., 1995, 50(9):1373-1382.
[45] L'vov B V. Mechanism of thermal decomposition of alkaline-earth carbonates [J].Thermochim Acta., 1997, 303(2):161-170.
[46] Broda M, Pacciani R, Müller C R. CO2CaptureviaCyclic Calcination and Carbonation Reactions//Porous Materials for Carbon Dioxide Capture [M]. Springer, 2014:181-222.
[47] Stanmore B R, Gilot P. Review—calcination and carbonation of limestone during thermal cycling for CO2sequestration [J].Fuel Process. Technol., 2005, 86(16):1707-1743.
[48] Feng Yun (冯云), Chen Yaxin (陈延信). Development of research on calcium carbonate for decomposed kinetics [J].Bulletin of theChinese Ceramic Society (硅酸盐通报),2006, (3):140-145.
[49] Martinez I, Grasa G, Murillo R, Arias B, Abanades J C. Kinetics of calcination of partially carbonated particles in a Ca-looping system for CO2capture [J].Energy Fuels, 2012, 26(2):1432-1440.
[50] Cremer E, Nitsch W. The function of CO2pressure on CaCO3decomposition rate [J].Z. Elektrochem., 1962, 66(8/9):697-702.
[51] Sharp J H, Wentworth S A. Kinetic analysis of thermogravimetric data [J].Analytical Chemistry, 1969, 41(14):2060-2062.
[52] Xie Jianyun (谢建云), Fu Weibiao (傅维标). Uniform mathematical model for limestone calcination [J].Journal of Combustion Science and Technology(燃烧科学与技术), 2002, 8(3):270-274.
[53] Ning Jingtao (宁静涛), Zhong Beijing (钟北京), Fu Weibiao (傅维标). Study on the calcination of fine limestone powder at high temperature [J].Journal of Combustion Science and Technology(燃烧科学与技术), 2003, 9(3):205-208.
[54] Ar I, Doğu G. Calcination kinetics of high purity limestones [J].Chem.Eng. Sci., 2001, 83(2):131-137.
[55] Milne C R, Silcox G D, Pershing D W, Kirchgessner, David A.Calcination and sintering models for application to high-temperature,short-time sulfation of calcium-based sorbents [J].Ind. Eng. Chem.Res., 1990, 29(2):139-149.
[56] Shi Qi (师琦), Wu Sufang (吴素芳), Jiang Mingzhe (蒋明哲), Li Qinghui (李清辉). Reactive sorption-decomposition kinetics of nano Ca-based CO2sorbents [J].CIESCJournal(化工学报),2009, 60(3):641-648.
[57] Dennis J S, Hayhurst A N. The effect of CO2on the kinetics and extent of calcination of limestone and dolomite particles in fluidised beds [J].Chem. Eng. Sci., 1987, 42(10):2361-2372.
猜你喜欢
陶瓷学报(2021年5期)2021-11-22 06:35:40
陶瓷学报(2021年5期)2021-11-22 06:35:36
中国食用菌(2021年10期)2021-11-04 06:23:20
陶瓷学报(2019年6期)2019-10-27 01:18:16
北京航空航天大学学报(2017年2期)2017-11-24 05:24:51
浙江大学学报(工学版)(2016年2期)2016-06-05 09:20:51
特产研究(2016年3期)2016-04-12 07:16:20
天津科技大学学报(2015年4期)2015-04-16 04:55:09
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01 02:54:19
石油化工应用(2014年6期)2014-03-11 17:39:38
