
溶液燃烧法制备的Ni基催化剂及其浆态床甲烷化催化性能
2015-04-01 06:16吉可明孟凡会高源李忠
无机化学学报 2015年2期
吉可明 孟凡会 高源 李忠
(太原理工大学,煤科学与技术教育部和山西省重点实验室,太原030024)
溶液燃烧法制备的Ni基催化剂及其浆态床甲烷化催化性能
吉可明 孟凡会 高源 李忠*
(太原理工大学,煤科学与技术教育部和山西省重点实验室,太原030024)
分别以硝酸铝、硝酸氧锆、硝酸镧和硝酸铈为载体前驱体,与硝酸镍和尿素配制水溶液,采用溶液燃烧法制备了Ni-Al2O3、Ni-ZrO2、Ni-La2O3和Ni-CeO2催化剂,研究了浆态床CO甲烷化催化性能,并进行了低温N2吸附-脱附、XRD、SEM、TEM、H2-TPR和H2化学吸附等表征分析。结果表明,以硝酸铝为前驱体制备Ni-Al2O3催化剂时燃烧火焰稳定且持续时间长,达23 s,样品比表面积(468 m2·g-1)和金属Ni表面积(10 m2·g-1)均较大、Ni粒径小(3~5 nm)且分散度高,CO甲烷化催化活性和稳定性好,CO转化率和CH4选择性分别达到94%和95%,在100 h的甲烷化反应中未出现明显失活;以硝酸氧锆和硝酸镧为前驱体制备样品时未出现明显的燃烧火焰,持续时间仅为12 s和5 s,催化剂比表面积、金属表面积及催化活性均较低;以硝酸铈为前驱体制备样品时燃烧过程迅速而剧烈,样品比表面积(22 m2·g-1)和金属Ni表面积(5 m2·g-1)小、Ni粒径大且分散性差,甲烷化催化性能最差,CO转化率仅为41%,CH4选择性仅为89%。
催化;催化剂制备;浆态床反应器;CO甲烷化;溶液燃烧法;镍催化剂;载体
0 前言
煤气化制天然气是煤炭清洁利用的重要途径之一,其中CO加氢甲烷化是核心技术[1-2]。甲烷化反应是强放热反应[3],由于传热性差,固定床反应器易导致催化剂过热烧结或积炭。采用浆态床反应器能及时移除反应热,并可以使用富CO反应气提高单程转化率[4-5]。
甲烷化催化剂一般选用催化活性较好且价格低廉的Ni为活性组分,载体以Al2O3、ZrO2、La2O3或CeO2等金属氧化物为主[6-9]。研究发现,工业甲烷化催化剂和传统浸渍法制备的Ni催化剂在浆态床反应器中催化活性较低[10-11]。近年来,采用溶液燃烧法制备催化剂的研究日益增加[12-15],该法制备的催化剂具有比表面积大、颗粒小且活性金属组分分散均匀等特点[16]。Pfeil等[12]采用Co(NO3)2为前驱体,甘氨酸为燃料,制备得到蜂窝状泡沫结构的纳米Co3O4材料,比表面积达92 m2·g-1,催化NaBH4水解的效果比商业Co3O4催化剂提高约40%。Dinka等[13]采用Fe(NO3)3作为活性组分前驱体,甘氨酸为燃料,并加入Al2O3(或ZrO2)作为载体,通过溶液燃烧制备的Fe2O3/Al2O3(或Fe2O3/ZrO2)催化剂活性组分分散均匀,在重质烃类自热重整反应中12 h内总碳转化率接近100%。Sharma等[14]采用RuCl3和(NH4)2Ce(NO3)6为前驱体,草酸二酰肼为燃料,制备了Ru-CeO2催化剂用于CO2甲烷化反应,CO2转化率比浸渍法制备的催化剂提高约30%,CH4选择性达99%。Colussi等[15]采用Pd(NO3)2和(NH4)2Ce(NO3)6(或Al(NO3)3)为前驱体、草酸二酰肼为燃料,制备出Pd-CeO2(或Pd-Al2O3)催化剂,其中Pd-CeO2催化剂因Pd和Ce相互作用较强、Pd分散性好且晶粒小,在丙烷和二甲醚的催化燃烧反应中催化反应速率高于Pd-Al2O3催化剂,且达到浸渍法制备的Pd/CeO2催化剂的2倍以上。然而,以不同金属硝酸盐为前驱体,并采用溶液燃烧法制备Ni催化剂进而研究其浆态床CO甲烷化催化性能的报道较少。
本工作采用硝酸铝、硝酸氧锆、硝酸镧和硝酸铈为载体前驱体,与硝酸镍和尿素共燃烧制备了Ni-Al2O3、Ni-ZrO2、Ni-La2O3和Ni-CeO2催化剂,并对其进行了低温N2吸附-脱附、XRD、SEM、TEM、H2-TPR和H2化学吸附等表征,探讨了硝酸盐前驱体对催化剂微观结构及催化浆态床CO甲烷化性能的影响。
1 实验部分
1.1 催化剂制备
实验所用试剂:硝酸镍(上海国药,>98%),硝酸铝(天津光复,>99%),硝酸氧锆(天津光复,>99%),硝酸镧(天津光复,>99%),硝酸铈(天津光复,>99%),尿素(天津风船,>99%)。
根据溶液燃烧法投料计算方法[17],按照总氧化价和总还原价化学计量比1∶1配制硝酸镍、硝酸铝和尿素的水溶液,然后放入300℃的马弗炉中,溶液逐步浓缩,然后自行燃烧,待燃烧自动熄灭后,冷却并收集固体产物,记为Ni-Al2O3催化剂。以同样方法制备Ni-ZrO2、Ni-La2O3和Ni-CeO2催化剂。作为对比,先采用溶液燃烧制备γ-Al2O3(比表面积295 m2·g-1),然后浸渍一定量的硝酸镍,并经过450℃焙烧后得到Ni/Al2O3催化剂。以上所有样品金属Ni质量百分含量均为25%。
1.2 催化剂活性评价
将3 g催化剂放置在固定床还原装置中采用VH2/VN2=1/4还原气,气体流速125 mL·min-1,在550℃常压还原4 h。将还原后的2 g催化剂和120 mL液体石蜡放入250 mL高压反应釜中,反应温度280℃,反应压力1.0 MPa,原料气为VH2/VCO=3/1,气体体积空速为3 000 mL·g-1·h-1,反应釜搅拌速率为750 r·min-1。反应釜排出的气体经过5℃的冷水冷凝脱除反应生成水,然后将不凝性气体通过自动取样阀取样。在三阀四柱气相色谱(Aglient 7890A)在线分析组成,以He为载气,采用毛细管HP-AL/S柱(30 m×530 μm×15 μm)和氢火焰离子检测器(FID)分析C1~4烃类;以He为载气,填充柱PORAPAK Q为预分离柱,采用毛细管柱HP-PLOT/Q(30 m×530 μm×40 μm)、HP-MOLESIEVE(30 m×530 μm×25 μm)和热导率检测器(TCD)分析CO、CO2、CH4和N2等气体。采用标准气体为参照物,外标法计算尾气中各组分的含量。
1.3 催化剂表征
N2吸附采用3H-2000PS2型静态容量法比表面及孔径分析仪进行,样品在30 Pa,130℃脱气1 h。在液氮温度吸附N2,用BET公式计算比表面积。
X射线衍射(XRD)采用DX-2007型X射线衍射仪,Cu靶Kα射线(λ=0.154 18 nm),石墨单色器,管电压和管电流分别为40 kV和30 mA,扫描速度8° ·min-1,扫描范围10°~85°。
扫描电镜SEM采用JSM-6700F型扫描电子显微镜,最高加速电压30 kV,将样品粉末粘附于导电胶上制样。
透射电镜TEM采用GG314-JEM-2100F场发射透射电子显微镜,加速电压200 kV,将样品分散在乙醇中超声10 min后,将悬浮液滴在铜网上制样。
程序升温还原H2-TPR采用AutochemⅡ2920型全自动程序升温化学吸附仪进行分析。将约20 mg样品置于U型石英反应管中,通入Ar气,流速为50 mL·min-1,以10℃·min-1升温至350℃,恒温吹扫40 min,降温至100℃,切换VH2/VAr=1/9混合气体,流速50 mL·min-1,待基线稳定后,以10℃· min-1升温至800℃,H2的消耗用TCD检测。
H2化学吸附采用AutochemⅡ2920型全自动程序升温化学吸附仪。将约500 mg样品置于U型石英反应管中,通入VH2/VAr=1/9气氛,流速50 mL· min-1,10℃·min-1的升温速率升温至550℃,恒温还原4 h,降温至120℃,Ar气氛吹扫1 h,降温至50℃,基线平稳后,每3 min进行一次VH2/VAr=1/9气氛的脉冲吸附,进行15次脉冲,脉冲吸附信号用TCD检测。根据H2吸附量结果分别计算得到催化剂金属Ni分散度(D)和金属Ni表面积(SNi,m2·g-1),其计算公式如下:
祝国寺把一些传统文化和地方文化引入寺庙的做法,很有创意。记者注意到,这里不仅体现了中国浓郁的孝道文化,还融入了东川一些优秀的地方文化。寺庙的外墙上,连片地挂着阿旺草山的摄影作品,把寺庙装点成一个户外的摄影展厅。

式(1~2)中物理量表示如下:Q,H2吸附量,mmol·g-1;MNi,Ni相对原子质量,58.71;x,催化剂Ni含量,%;N0,阿伏伽德罗常数,6.02×1023;SA,Ni原子横截面积,0.064 9 nm2。
2 结果与讨论
2.1 催化剂共燃烧过程分析
对硝酸铝、硝酸氧锆、硝酸镧和硝酸铈四种金属硝酸盐与硝酸镍和尿素共燃烧制备催化剂的过程进行观察,如图1所示。由图1(a)可以看出,以硝酸铝为前驱体的样品出现了明显的起泡现象,随后出现烟雾及明亮的火焰,火焰在粘稠液体之间蔓延燃烧,燃烧过程平缓连续,时间长达23 s,产物呈疏松的棉花状。图1(b)和图1(c)分别为以硝酸氧锆和硝酸镧为前驱体制备催化剂过程,其样品燃烧不充分,属无焰阴燃[18],其中以硝酸氧锆为前驱体的样品燃烧过程释放大量烟雾,持续时间12 s,产物为多孔块状固体,极易破碎;以硝酸镧为前驱体的样品燃烧过程短暂并伴有烟雾释放,用时仅5 s,产物为致密颗粒,放在空气中极易潮解。图1(d)以硝酸铈为前驱体制备样品时,燃烧过程迅速且剧烈,用时仅4 s,产物为致密颗粒。
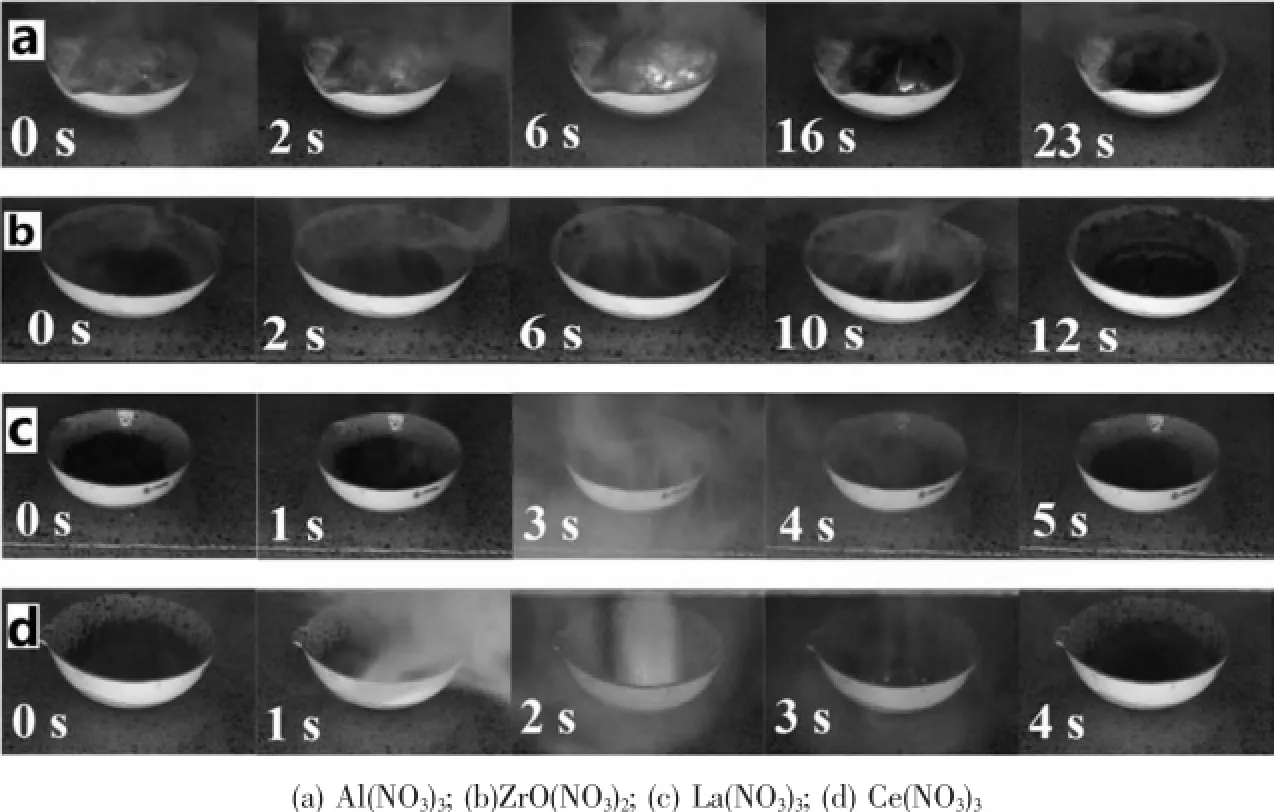
图1 不同前驱体制备催化剂过程的照片Fig.1Images of catalysts prepared with different precusors
2.2 催化剂的N2吸附表征
不同催化剂还原前和还原后的N2吸附表征结果见表1。可以看出,不同载体前驱体制备的催化剂比表面积差异很大,其中Ni-Al2O3催化剂的比表面积最大,达到468 m2·g-1,这是由于采用硝酸铝为前驱体制备催化剂时,样品燃烧持续时间长,且燃烧过程中形成的大量孔道有利于增大产物的比表面积;Ni-ZrO2催化剂比表面积为150 m2·g-1;Ni-La2O3催化剂燃烧过程持续时间短,比表面积为54 m2·g-1;Ni-CeO2催化剂燃烧过程短暂而剧烈,容易引起产物的高温烧结,燃烧后制备的催化剂比表面积仅为22 m2·g-1。从表中还可以看出,所有催化剂经550℃还原后,比表面积和孔容均出现不同程度下降,其中Ni-ZrO2催化剂比表面积下降48%,Ni-La2O3比表面积下降39%。表中Ni-La2O3催化剂的孔径由还原前的16.0 nm降为13.2 nm,其他催化剂的孔径则介于2.2~3.6 nm之间,还原前后变化不大。
2.3 催化剂的XRD表征
不同前驱体制备的催化剂还原前及还原后的XRD图见图2。从图2(a)中可以看出,Ni-Al2O3催化剂出现了γ-Al2O3(PDF No.10-0425)的特征衍射峰,未检测到明显的NiO(PDF No.65-5745)特征衍射峰,表明Ni物种分散均匀。Ni-ZrO2催化剂出现NiO的衍射峰。Ni-La2O3催化剂出现了LaONO3(PDF No.31-0665)的特征衍射峰,表明在溶液燃烧过程中硝酸镧热分解为LaONO3,这是Ni-La2O3催化剂在空气中很容易潮解的原因[19],图中没有观察到Ni物种的衍射峰。Ni-CeO2催化剂存在明显的CeO2(PDF No.34-0394)和NiO特征衍射峰,表明硝酸镍和硝酸铝在溶液燃烧过程中热分解为CeO2和NiO晶体。
从图2(b)可以看出,还原后的Ni-Al2O3催化剂出现了微弱的金属Ni(PDF No.65-2865)特征衍射峰;Ni-ZrO2催化剂还原后出现了金属Ni和Zr2O (PDF No.65-0461)的特征衍射峰;Ni-La2O3催化剂中仍然存在LaONO3衍射峰,未发现金属Ni的特征衍射峰;还原后的Ni-CeO2催化剂仍然出现了CeO2的衍射峰,并检测到强度较弱的金属Ni衍射峰。
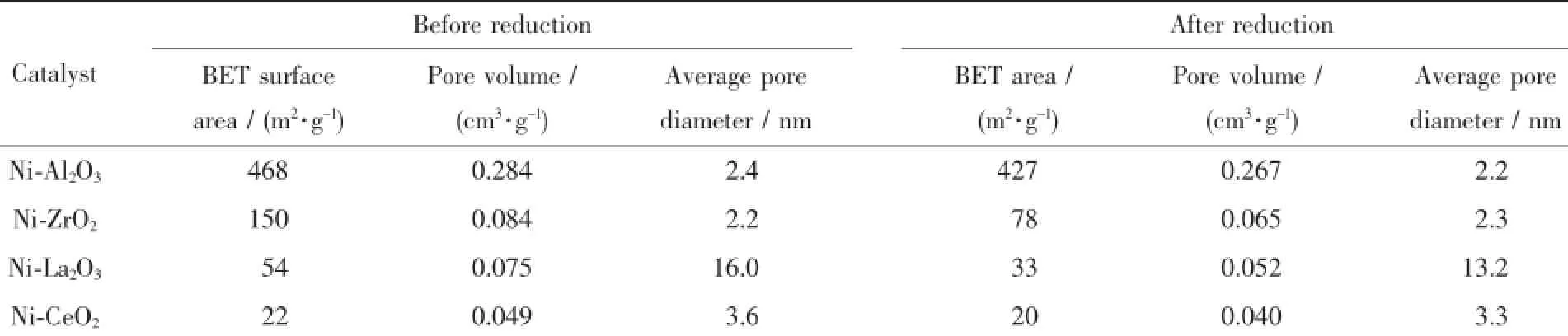
表1 不同催化剂还原前及还原后的织构性质Table 1Textural property of different catalysts before and after reduction
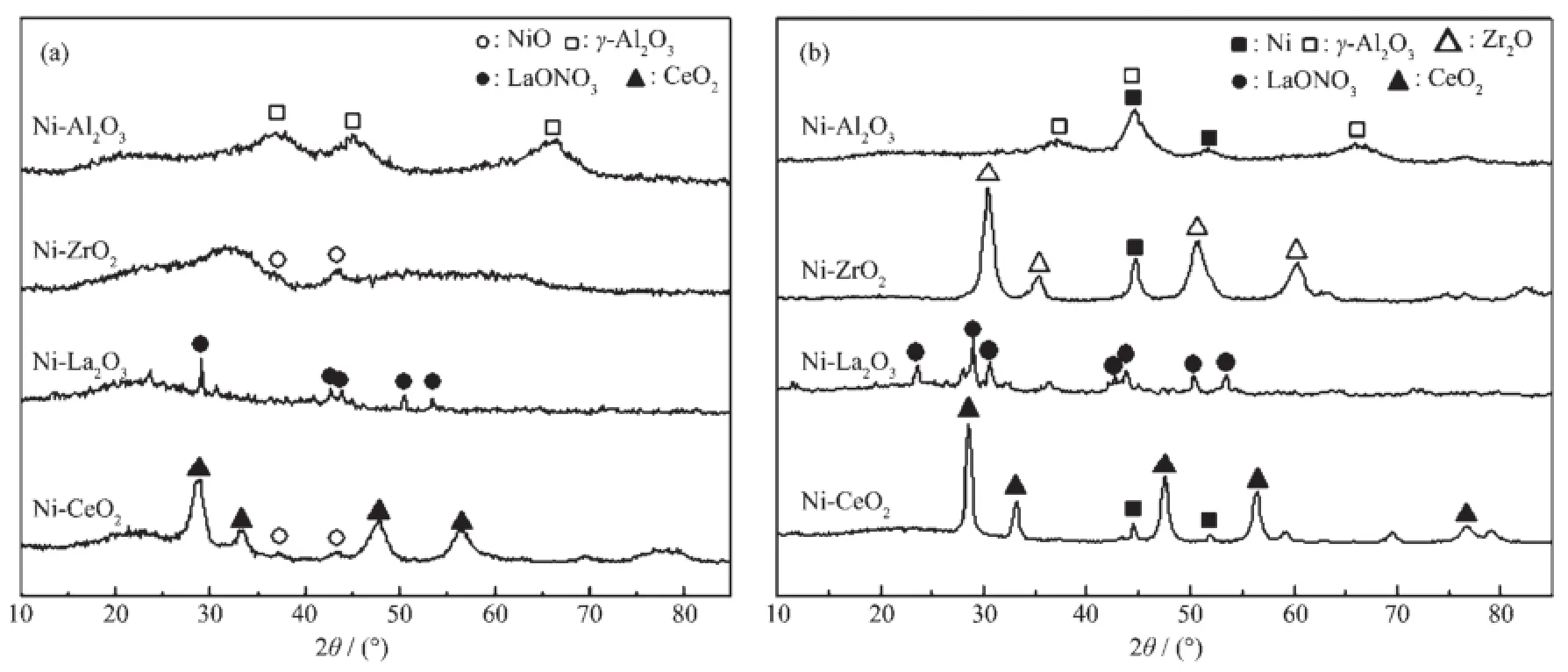
图2 不同催化剂还原前(a)和还原后(b)的XRD图Fig.2XRD patterns of different catalysts before(a)and after(b)reduction
2.4 催化剂的形貌表征
催化剂还原前的SEM表征图片示于图3。从图中可以看出,Ni-Al2O3催化剂为不规则的球状颗粒,不同尺寸的颗粒堆积聚集后形成较大疏松状的颗粒;Ni-ZrO2催化剂形貌与Ni-Al2O3催化剂相差不大,但Ni-ZrO2催化剂表面的小颗粒堆积形成较大的团聚体;Ni-La2O3催化剂燃烧过程中形成LaONO3,因而样品容易潮解,样品表面出现不规则形状的空隙;Ni-CeO2催化剂样品表面致密,为较大的块状。
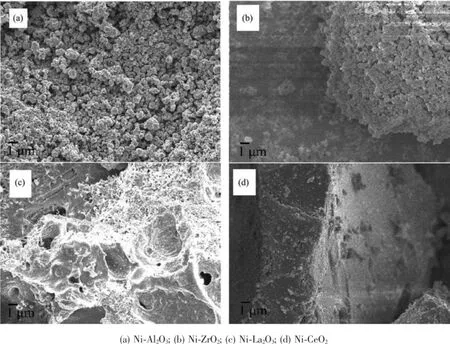
图3 催化剂的SEM图Fig.3SEM images of catalysts

图4 催化剂的TEM图Fig.4TEM images of catalysts
催化剂还原前的TEM表征图片示于图4。从图中可以看出,Ni-Al2O3催化剂上NiO晶粒尺寸约3~5 nm,晶粒小且分散均匀;Ni-ZrO2催化剂中NiO晶粒较大,约10 nm左右;Ni-La2O3催化剂质地均匀,局部放大后可以观察到粒径约2~3 nm的NiO粒子;Ni-CeO2催化剂也具有10 nm左右的较大的NiO粒子,这是由于燃烧过程短暂而剧烈,催化剂的晶粒烧结长大而成。
2.5 催化剂的H2-TPR表征
不同催化剂的H2-TPR表征结果见图5。由图可以看出,Ni-Al2O3催化剂在280℃和480℃左右出现了较强的低温和高温还原峰,分别对应于自由态NiO和分散态NiO的还原,其中自由态NiO与载体无相互作用或相互作用较弱[20-22],还原后形成的Ni粒子较大[23],而分散态NiO与载体的相互作用较强,还原后颗粒较小,是催化反应中的主要活性物种[23],从图中可以看出,Ni-Al2O3催化剂的高温峰峰面积较大,表明Ni物种大多均匀分散在Al2O3载体上。Ni-ZrO2催化剂在270℃和430℃也出现了自由态和分散态NiO的还原峰,相比于Ni-Al2O3催化剂,Ni-ZrO2催化剂的2个还原峰均向低温方向偏移,归因于Ni物种与载体的相互作用较弱。Ni-La2O3催化剂在260℃也出现了低温还原峰,高温峰出现在500℃左右。Ni-CeO2催化剂仅在270oC出现1个低温还原峰,这是由于Ni-CeO2催化剂比表面积小,Ni物种主要以自由态NiO形式存在,因而与载体相互作用弱,催化剂还原温度低。
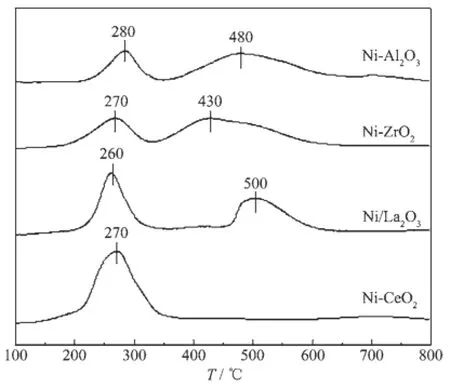
图5 不同催化剂的H2-TPR图Fig.5H2-TPR profiles of different catalysts
2.6 催化剂的H2化学吸附表征
不同催化剂还原后的H2化学吸附结果列于表2。由表可见,Ni-Al2O3催化剂的金属表面积和金属分散度最大,分别为10 m2·g-1和8%,这是由于Ni-Al2O3催化剂比表面积大,有利于活性组分Ni的分散,从而有利于提供更多的催化活性位并进而提高催化剂甲烷化活性。Ni-ZrO2和Ni-La2O3催化剂的金属表面积和金属分散度居中。Ni-CeO2催化剂的金属表面积和金属分散度最小,分别为5 m2·g-1和4%,这是由于Ni-CeO2催化剂比表面积小,金属Ni分散性差。金属表面积和金属分散度按递减顺序依次为Ni-Al2O3>Ni-ZrO2>Ni-La2O3>Ni-CeO2。2.7催化剂的浆态床甲烷化催化性能

表2 不同催化剂还原后的H2化学吸附数据Table 2Data of H2-chemisorption of different catalysts
不同催化剂的浆态床CO甲烷化催化性能见表3,表3同时列出了浸渍法制备的Ni/Al2O3催化剂的催化结果。表中所有催化剂在20 h内的评价过程中未出现明显失活,所有数据均取自催化剂活性数据的平均值。从表中可以看出,Ni-Al2O3催化剂的CO甲烷化催化性能最佳,CO转化率和CH4选择性分别达到94%和95%,CH4产率达到90%。作为对比,浸渍法制备的Ni/Al2O3催化剂的CO转化率和CH4选择性分别为86%和92%,CH4产率仅为79%,明显低于Ni-Al2O3催化剂的CH4产率。这是由于Ni-Al2O3催化剂比表面积大,金属Ni在载体上分散性好,催化活性位多,因而有利于甲烷化反应的进行。Ni-ZrO2催化剂甲烷化性能次之,其CO转化率和CH4产率分别为86%和76%。Ni-CeO2催化剂甲烷化性能最差,其CO转化率和CH4产率仅为41%和37%。从表中还可以看出,Ni-Al2O3催化剂的副产物C2-C4和CO2选择性仅为1%和4%,明显低于其他催化剂,这是由于Zr、La和Ce能够促进水汽变换反应,从而在反应中形成较多的CO2[24-26]。表中不同催化剂的甲烷化催化性能按递减顺序依次为Ni-Al2O3>Ni-ZrO2>Ni-La2O3>Ni-CeO2。
Ni-Al2O3催化剂100 h的浆态床甲烷化性能评价结果如图6所示。从图中可以看出,Ni-Al2O3催化剂在100 h的反应时间CO转化率和CH4、CO2、C2-C4烃类选择性没有显著变化,催化剂稳定性较好。从TEM和H2-TPR表征结果可以看出,催化剂Ni物种分散较好且与载体相互作用较强,导致其催化稳定性较好。
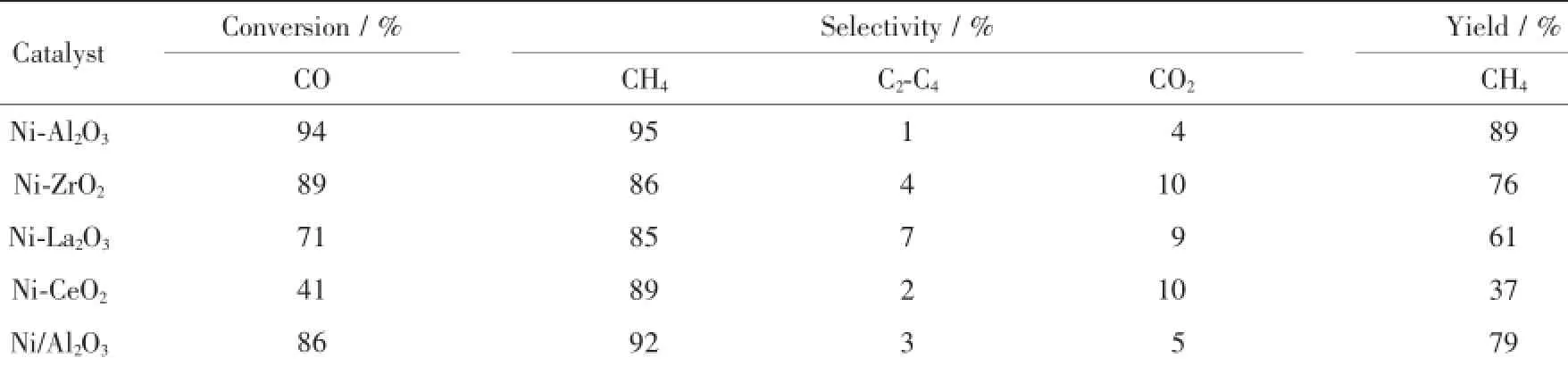
表3 不同Ni基催化剂的浆态床CO甲烷化催化性能Table 3Catalytic performance of different Nickel-based catalysts for slurry CO methanation
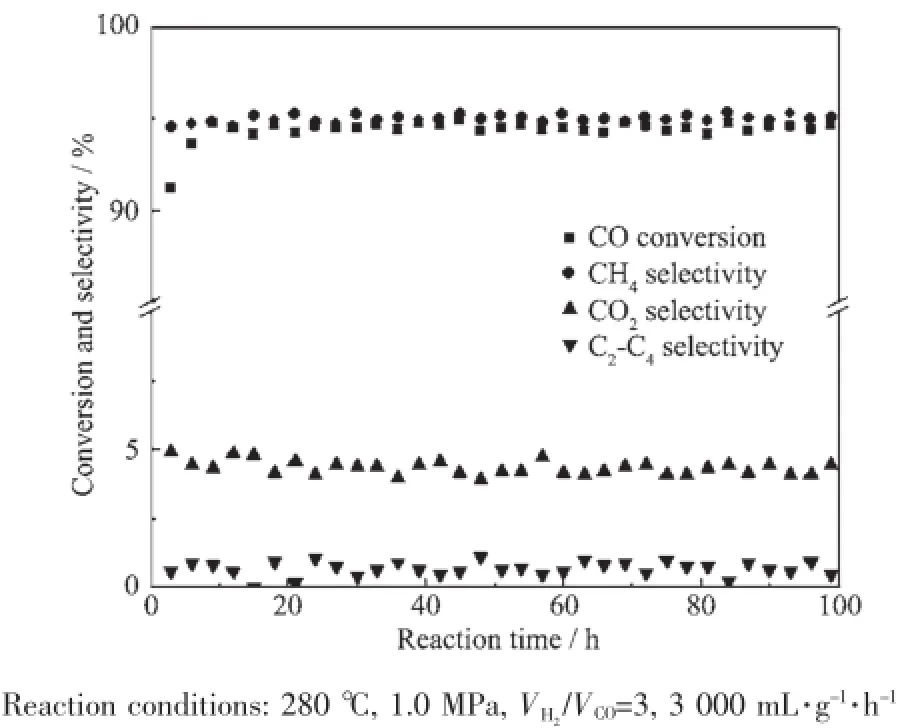
图6 Ni-Al2O3催化剂在浆态床甲烷化反应中的稳定性Fig.6Stability of Ni-Al2O3catalyst for slurry CO methanation
3 结论
以硝酸铝为前驱体制备Ni-Al2O3催化剂时燃烧持续时间长,样品比表面积和金属Ni表面积大、Ni粒径小且分散均匀;以硝酸氧锆和硝酸镧为前驱体制备样品时未出现明显火焰,燃烧持续时间短,催化剂比表面积和催化活性居中;以硝酸铈为前驱体制备样品时燃烧过程迅速而剧烈,样品比表面积和金属Ni表面积小、Ni粒径大且分散性差。各催化剂的浆态床CO甲烷化催化性能顺序为Ni-Al2O3>Ni-ZrO2>Ni-La2O3>Ni-CeO2,Ni-Al2O3催化剂的甲烷化性能最佳,CO转化率和CH4选择性分别达到94%和95%,优于浸渍法制备的Ni/Al2O3催化剂,并且在100 h的甲烷化反应中未出现明显失活。
[1]Kopyscinski J,Schildhauer T J,Biollaz S M A.Fuel,2010,89 (8):1763-1783
[2]Zhang J,Bai Y,Zhang Q,et al.Fuel,2014,132:211-218
[3]Gao J,Wang Y,Ping Y,et al.RSC Adv.,2012,2(6):2358-2368
[4]CUI Xiao-Xi(崔晓曦),FAN Hui(范辉),ZHENG Hua-Yan(郑华艳),et al.Chinese J.Inorg.Chem.(无机化学学报),2012, 28(3):495-502
[5]CUI Xiao-Xi(崔晓曦),MENG Fan-Hui(孟凡会),HE Zhong (何忠),et al.Chinese J.Inorg.Chem.(无机化学学报),2013, 30(2):277-283
[6]Zhao A,Ying W,Zhang H,et al.Catal.Comun.,2012,17: 34-38
[7]Tada S,Shimizu T,Kameyama H,et al.Int.J.Hydrogen Energy,2012,37(7):5527-5531
[8]Cai M,Wen J,Chu W,et al.J.Nat.Gas Chem.,2011,20 (3):318-324
[9]SONG Huan-Ling(宋焕玲),YANG Jian(杨建),ZHAO Jun(赵军),et al.Chin.J.Catal.(催化学报),2010,31(1):21-23
[10]Meng F H,Zhong P Z,Li Z,et al.J.Chem,2014,2014:1-7
[11]MENG Fan-Hui(孟凡会),LIU Jun(刘军),LI Zhong(李忠), et al.J.Fuel Chem.Technol.(燃料化学学报),2014,42(2): 231-237
[12]Pfeil T L,Pourpoint T L,Groven L J.Int.J.Hydrogen Energy,2014,39(5):2149-2159
[13]Dinka P,Mukasyan A S.J.Phys.Chem.B,2005,109(46): 21627-21633
[14]Sharma S,Hu Z,Zhang P,et al.J.Catal.,2011,278(2):297-309
[15]Colussi S,Gayen A,Llorca J,et al.Ind.Eng.Chem.Res., 2012,51(22):7510-7517
[16]González-Cortés S L,Imbert F E.Appl.Catal.A,2013,452: 117-131
[17]Jung C H,Jalota S,Bhaduri S B.Mater.Lett.,2005,59(19/ 20):2426-2432
[18]LU Chang(路长),ZHOU Jian-Jun(周建军),LIN Qi-Zhao(林其钊),et al.J.Combust.Sci.Technol.(燃烧科学与技术), 2005,11(1):41-46
[19]WU Shu-Rong(吴淑荣),XIONG Wei-Miao(熊为淼),HE Ming-An(何明安),et al.J.Northwest Univ.(西北大学学报),1981,22(3):32-38
[20]Zou X,Wang X,Li L,et al.Int.J.Hydrogen Energy,2010, 35(22):12191-12200
[21]Yang J,Wang X,Li L,et al.Appl.Catal.B,2010,96(1-2): 232-237
[22]Koo K Y,Roh H-S,Seo Y T,et al.Int.J.Hydrogen Energy, 2008,33(8):2036-2043
[23]Zhang J,Xu H,Jin X,et al.Appl.Catal.A,2005,290(1-2): 87-96
[24]Nagai M,Zahidul A M,Kunisaki Y,et al.Appl.Catal.A, 2010,383(1-2):58-65
[25]Kam R,Selomulya C,Amal R,et al.J.Catal.,2010,273(1): 73-81
[26]LIU Na(刘娜),DU Xia-Ru(杜霞茹),YUAN Zhong-Shan(袁中山),et al.J.Chinese Soc.Rare Earths(中国稀土学报), 2005,23(1):26-30
Solution Combustion Prepared Ni-Based Catalysts and Their Catalytic Performance for Slurry Methanation
JI Ke-MingMENG Fan-HuiGAO YuanLI Zhong*
(Key Laboratory of Coal Science and Technology of Ministry of Education and Shanxi Province, Taiyuan University of Technology,Taiyuan 030024,China)
Ni-Al2O3,Ni-ZrO2,Ni-La2O3and Ni-CeO2catalysts were prepared by solution combustion method using Al(NO3)3,ZrO(NO3)2,La(NO3)3and Ce(NO3)3(mixed with Ni(NO3)2and urea in aqueous solution)as the support precursor,respectively.The CO methanation performances of catalysts were studied in slurry-bed reactor,and the catalysts were characterized by low temperature N2adsorption-desorption,XRD,SEM,TEM,H2-TPR and H2chemsorption.The results show that the combustion preparation process of Ni-Al2O3catalyst using Al(NO3)2as the precursor is stable for long-duration(up to 23 s)and the catalyst has larger surface area(468 m2·g-1)and metal surface area(10 m2·g-1),smaller Ni particle(3~5 nm),excellent dispersion of Ni,and the catalyst has good catalytic performance,whose CO conversion and CH4selectivity are 94%and 95%,respectively,and no catalyst deactivation is observed in 100 h.The preparation process for catalysts using ZrO(NO3)2and La(NO3)3as precursors does not show obvious flame and burning time is also shorter(12 s and 5 s),the surface areas,metal surface areas and catalytic performances are lower than that of Ni-Al2O3while that for the catalyst using Ce(NO3)2as the precursor has high intensity combustion.The catalyst obtained from Ce(NO3)2precursor shows lower surface area(22 m2·g-1)and metal surface area(5 m2·g-1),larger Ni particle and worse dispersion of Ni and theworst methanation catalytic performance with CO conversion and CH4selectivity of 41%and 89%,respectively.
catalysis;catalyst prepration;slurry-bed reactor;CO methanantion;solution combustion method;Ni-based catalyst; support
O614;O643.3
A
1001-4861(2015)02-0267-08
10.11862/CJIC.2015.050
2014-08-04。收修改稿日期:2014-11-07。
国家“973”计划(No.2012CB723105)、山西省青年基金(No.2013021007-4)、中国博士后科学基金(2013M541210)和太原理工大学校青年团队基金(No.2013T091)资助项目。
*通讯联系人。E-mail:lizhong@tyut.edu.cn,Tel/Fax:0351-6018526;会员登记号:S06N4167M1005。
猜你喜欢
军民两用技术与产品(2021年10期)2021-03-16
中学生数理化·高一版(2020年11期)2020-12-14
陶瓷学报(2020年2期)2020-10-27
水上消防(2020年1期)2020-07-24
中学化学(2019年2期)2019-07-08
疯狂英语·新读写(2018年3期)2018-11-29
中国资源综合利用(2017年4期)2018-01-22
当代化工研究(2016年7期)2016-03-20
中国资源综合利用(2016年6期)2016-01-22
应用化工(2014年11期)2014-08-16
