
杜氏肌营养不良症的表达谱分析
2015-03-29 13:24曹军华田礼军邓星强张传玲钱同宋贤响黄宝山
河北医药 2015年10期
曹军华 田礼军 邓星强 张传玲 钱同 宋贤响 黄宝山
独立行走能力[2],在20岁左右死于呼吸系统衰竭[3]。认知缺陷也是该病的特征之一[4]。虽然患者自出生起一直存在肌肉缺陷,但是DMD患者在出生的临床表征是正常的。DMD患者最初的症状多在2~5岁时有所表现,症状包括行走滞后于同龄人、经常摔倒、奔跑或者爬楼梯困难等[5,6]。因此,患者最初的两年被认为临床症状前阶段[7]。在DMD基因被发现以来的20多年内,研究者提出了很多有前景的治疗方法[8,9]。但是,这些方法有很多困难需要克服,目前该病仍无药可医。虽然DMD基因的突变是改变的首要原因,但是其他因素也可能与该病的病理过程相关。患者的次级变化涉及细胞因子[10]、一氧化氮合成酶[11]、代谢异常[12]、生长因子[13]、钙平衡[14]和肥大细胞脱颗粒作用[15]等过程,表明DMD的病理过程非常复杂,很多生化代谢通路可能与疾病进程相关。现有的高通量技术
杜氏进行性肌营养不良症(Duchenne muscular dystrophy,DMD)是一种因X染色体的肌营养不良蛋白基因发生突变而导致的以肌肉纤维化和中枢神经系统异常为特征的一种严重的进行性疾病。每年约有1/3 500的男性新生儿罹患DMD[1]。肌营养不良蛋白是一个427 kDa细胞骨架蛋白,位于肌纤维膜底部,与肌纤维膜蛋白共同构成肌营养不良蛋白相关蛋白复合物 (dystrophin-associatedproteincomplex,DAPC)。DAPC的缺失和缺陷会导致慢性炎症,进行性肌肉衰退和肌肉纤维化[2]。DMD患者通常在13岁之前丧失使得同时研究成千上万个基因的表达状况成为可能,为研究DMD的疾病发生机制提供了极大的便利。我们认为,异常的代谢通路与疾病的特定阶段相关。富集了差异表达基因的代谢通路可能在疾病进程过程起到重要作用。本研究中,我们对从基因表达数据库(gene expression omnibus,GEO)下载的两个表达谱芯片数据进行了分析,旨在鉴定出与发病阶段相关的代谢通路。此外,在不同发病阶段表达状况不同的基因分析也可能为以后的诊疗研究提供可靠的靶标。
1 材料与方法
1.1 芯片数据 我们从GEO数据库(http://www.ncbi.nlm.nih.gov/geo/)下 载 了 两 个 数 据 集 合(GSE6011 and GSE3307),共包含19名症状发生前的患者,8名症状发生后的患者和14名健康对照。两个数据集均基于GPL96平台,使用Affymetrix Human Genome U133A芯片。这些样本收集于2005至2006年间,由研究者在其文章发表后上传于GEO数据库。见表1。
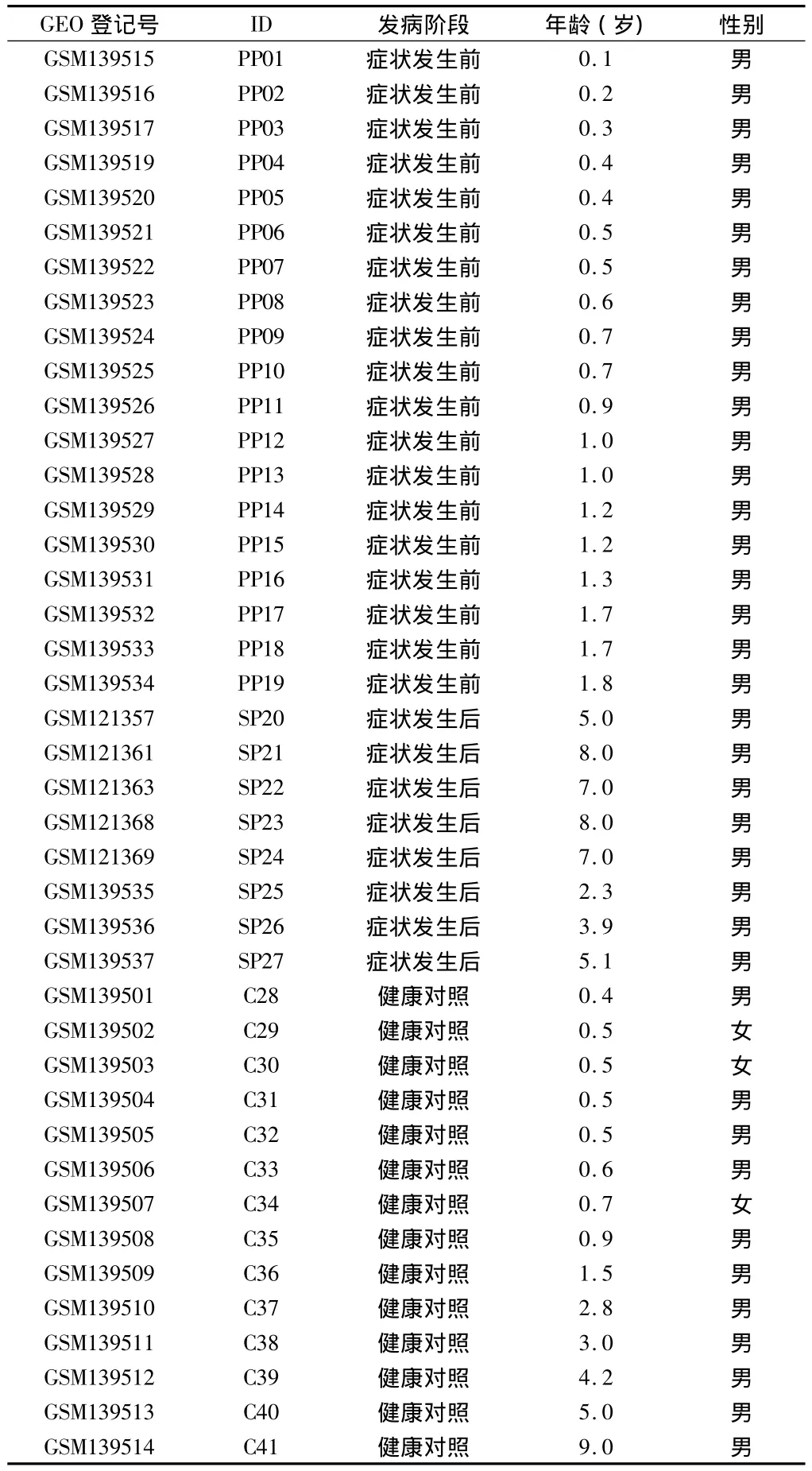
表1 样本信息
1.2 差异表达基因分析 我们下载了41个个体的CEL格式和SOFT格式的文件。原始数据采用multiarray analysis(RMA)[16]方法进行标准化,步骤如下:(1)去除背景噪音;(2)表达值标准化;(3)针对每个探针产生相应的表达值。t检验被用来检测在不同组中的差异表达基因(阈值0.01),Benjamini和 Hochberg法(Benjamini and Hochberg)被用来进行多重检验校正。与正常对照相比,症状发生前患者的差异表达基因和症状发生后患者的差异表达基因被检测出来。基因表达的上下调由表达量的差异确定。所有分析过程均在 R 软件(v2.14.1)中选用 BioConductor,limma 包(3.12.1)和库[17]。
1.3 通路富集分析 差异表达的探针通过SOFT格式文件进行注释。所有的基因均映射到KEGG通路数据库中 (http://www.genome.jp/kegg/)。我们采用超几何分布检验来检测存在差异表达基因富集的通路。
2 结果
2.1 症状发生前呈现差异表达基因富集的通路 与健康对照相比,在症状发生前的患者中,有1023个基因差异表达。其中,747个基因表达量上调,276个基因表达量下调。对于症状发生后的患者,1 407个基因差异表达,其中,977个基因表达量上调,430个基因表达量下调。共5 157个基因可以映射到KEGG数据库中。差异表达的基因中,症状发生前差异表达基因有494个可以映射到KEGG数据库,症状发生后差异表达的基因有619个可以映射到KEGG数据库中。富集分析的结果表明,在症状发生前的个体中,有41个通路富集了差异表达基因。在症状发生后的患者中,有33个通路富集了差异表达基因。在这些通路中,有11个通路仅在症状发生前发现,其中9个与代谢相关。见表2。

表2 仅在症状发生前呈现出差异表达基因富集的通路
2.2 症状发生前后均呈现差异表达基因富集的通路 有3个通路仅在症状发生后的患者中被鉴别出来,包含细菌入侵上皮细胞通路、紧密连接通路、D型谷氨酰胺和D型谷氨酸代谢通路。30个通路是两个阶段的患者共有的,其中包含两个DMD基因参与的通路,扩张型心肌病通路(hsa05414)和病毒性心肌炎通路(hsa05416)。见表3。
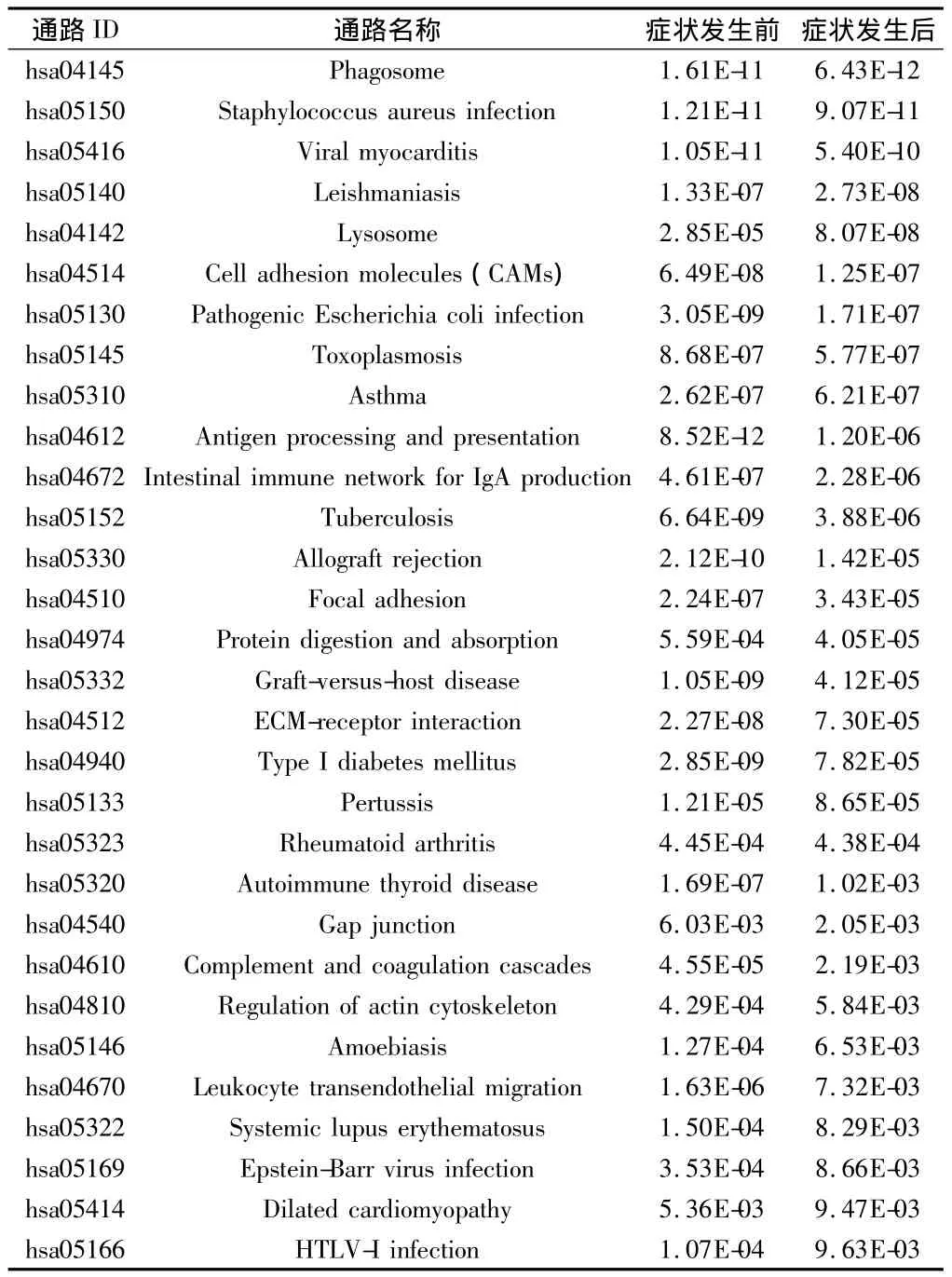
表3 在症状发生前后均呈现出差异表达基因富集的通路
2.3 相互作用 这些通路之间存在着相互关系,一些在不同阶段差异表达的基因。见图1、2。
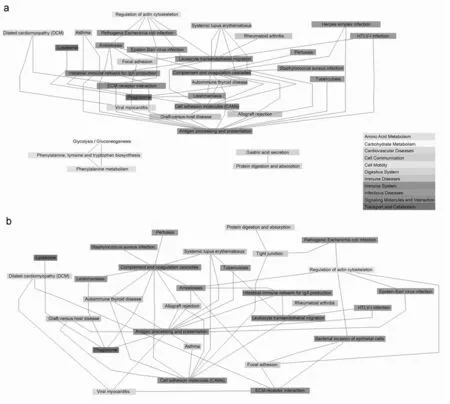
图1 差异表达基因富集的通路间的相互作用关系。a:症状发生前;b:症状发生后。仅显示KEGG数据库中与其他通路存在相互作用关系的通路
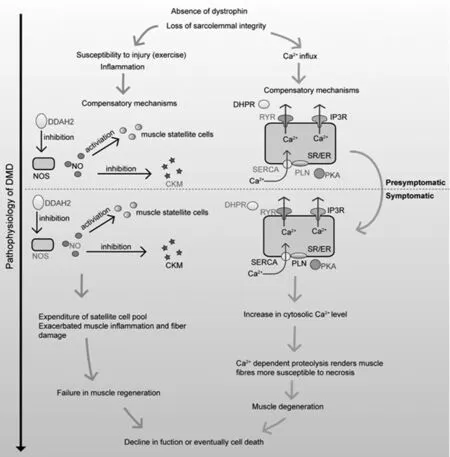
图2 DMD病理过程图示,仅展示了与肌肉重建和钙平衡维护相关的差异表达基因。由过表达的基因编码的蛋白质用红色显示,低表达的由绿色显示,黑色代表没有差异表达;DHPR是心肌特异性的,在心肌细胞中,RYR基因(RYR2)并没有差异表达
3 讨论
DMD的疾病进程比较复杂。因肌营养不良基因突变引起的次级过程包括肌纤维膜的破坏、钙平衡失调、肌肉坏死和衰竭[18],同时伴随着脂肪组织的增殖和免疫细胞的浸润等[3]。这些下游的通路可能与疾病进程相关,但是无法单独阐明DMD的发病进程。现有的芯片技术使得同时研究很多mRNA的差异表达成为可能。富集了差异表达基因的生化通路可能在DMD的发病进程中起到非常重要的作用。我们利用从GEO数据库下载的两套数据对不同发病阶段的差异表达基因和异常通路进行了分析。
结果表明,两个发病阶段共有超过70%的异常通路,表明很多通路异常在患者中是一直存在的,不论其是否表现出临床表型。如图1所示,很多通路设计免疫系统,包括炎症/免疫反应、免疫性疾病、免疫系统和传染病。这些免疫相关的通路的异常可能与免疫细胞的渗透相关,包含 T 细胞[19,20]、巨噬细胞[19,20]、嗜酸细胞[21]和肥大细胞[22,23]。这些免疫性细胞的渗透会导致很多炎性细胞因子水平的升高。以往的研究表明在小鼠模型中降低CD4或者 CD8 T cells[24]或者巨噬细胞[25]的水平可显著缓解小鼠的DMD表型,延缓其发病进程[26]。与以往的研究[27]一致,共有免疫或炎症相关的通路表明患者出生后不久炎症级联反应即已启动。但是,有些基因在症状发生后表现出比症状发生前更高或者更低的差异表达,这些差异可能与其临床表现相关。
因肌营养不良蛋白缺失而导致的肌纤维膜完整性的丧失会知道肌肉对损伤的敏感性增高,导致炎症发生,同时启动肌肉重建过程。另外,以往的研究[3,18,28-30]表明细胞外钙离子的流入可能会导致随后的肌肉坏死和细胞凋亡。因此,我们针对肌肉重建过程和钙离子平衡维护过程中的基因的表达情况进行了进一步分析。
肌肉重建过程中,纤维化的进程可能在两个阶段差异不大。主要的纤维形成胶原蛋白(Ⅰ型和Ⅲ)和其他纤维胶原(Ⅴ型)在两个阶段均呈现出过表达。但是,如图2所示,NOS1的差异表达仅在症状发生后的病人中检测到。该基因的低表达可能会导致一氧化氮自由基的表达降低,而一氧化氮自由基可以保护肌肉免受氧化损伤[31],并且可激活肌肉卫星细胞[32]。以往的研究表明,在肌营养不良小鼠中维持正常水平的NOS,可以降低肌肉中巨噬细胞的浓度,从而起到预防肌肉膜损伤的作用,同时可降低肌酸激酶的浓度[25]。大量肌酸激酶的释放会引起相应的炎性反应。我们注意到肌肉肌酸激酶的活性在症状发生前的患者中表达水平是降低的,说明NOS在症状发生前可能功能是正常的,可激活正常的肌肉重建过程,预防进一步的肌肉损伤。因此,NOS的异常表达可能与DMD的疾病进程相关。此外,抑制NOS活性的二甲基精氨酸二甲基氨基水解酶2(dimethylarginine dimethylaminohydrolase 2,DDAH2)在两个阶段的患者中均呈现出过表达。但是过表达的水平在症状发生后(0.55~0.58)略高于症状发生前(0.37 ~0.46),这说明 NOS的表达水平降低可能与DDAH2的表达水平升高有关。
对钙平衡来说,随着细胞外钙离子通过肌营养不良蛋白缺陷膜的涌入,肌质网(sarcoplasmic reticulum,SR)钙泵细胞膜上的纳钙交换泵(sodium/calcium exchanger,NCX)和钙离子-ATP酶泵(Plasma membrane Ca2+-ATPase,PMCA)可起到降低胞内钙离子浓度的作用。我们的研究表明,与NCX和PMCA相关的基因均没有差异表达。但是,如图2所示,肌质/内质网钙腺苷三磷酸酶(sarcoplasmic/endoplasmic reticulum calcium ATPase,SERCA)和受磷蛋白 (phospholamban,PLN)在症状发生前表现出低表达。PLN/SERCA实际上降低了因为PLN的表达的降低量(-1.00419)大概是SERCA(ATP2A1:-0.50584;ATP2A2:-0.55769)的两倍。SERCA的活性受PLN的调控,在PLN去磷酸化时可以与SERCA相互作用从而抑制其活性[33]。蛋白激酶(protein kinase-A,PKA)可促使PLN磷酸化,从而取消对SERCA的抑制作用,增加其对钙离子的亲附性。PLN/SERCA比率的降低可促进SERCA对将钙离子吸收到内质网的作用[34]。因此,在症状发生前的患者中,PKA的过表达以及PLN/SERCA比率的降低可能会加速PLN的磷酸化,解除对hSERCA的抑制,促进钙离子从胞质进入内质网,从而降低胞质钙离子的浓度。而CACNA1D和CACNA1B基因仅在症状发生后的患者中呈现出低表达。他们所表达的蛋白是二氢吡啶受体(dihydropyridine receptors,DHPR)的亚基,可与阿诺碱受体(ryanodine receptors,RYR)结合并抑制内质网释放钙离子[35]。这两个基因的低表达可能会解除DHPR的一直作用,从而导致胞质钙离子浓度升高。在两个阶段的患者中,肌肉中的 RYR基因RYR1均呈现出低表达。但是SERCA对钙离子的亲和性可能在症状发生后降低因为PLN/SERCA的比率在症状发生后没有改变。因此,钙离子浓度的维持方面,症状发生前的患者可能优于症状发生后的患者。DHPR,PLN/SERCA可作为以后研究的靶点。
总之,我们针对DMD症状发生前患者和症状发生后患者的表达谱进行了分析以获得疾病进行过程中的作用机制。结果表明,两个阶段共有很多炎症相关的异常通路,说明疾病早期炎症级联反应已开始发生。另外,在两个阶段的特异性方面,我们发现症状发生前病人在肌重建过程和心肌钙平衡维持方面的表现优于症状发生后的患者。我们的研究为进一步了解DMD的疾病进程机制和以后的治疗方法研究奠定了基础。
1 Emery AE.The muscular dystrophies.Lancet,2002,359:687-695.
2 Hoffman EP,Brown RH,Kunkel LM.Dystrophin:the protein product of the Duchenne muscular dystrophy locus.Cell,1987,51:919-28.
3 Blake DJ,Weir A,Newey SE,et al.Function and genetics of dystrophin and dystrophin-related proteins in muscle.Physiol Rev,2002,82:291-329.
4 Anderson JL,Head SI,Rae C,et al.Brain function in Duchenne muscular dystrophy.Brain,2002,125(Pt 1):4-13.
5 Dubowitz V.Muscle disorders in childhood.Major Probl Clin Pediatr,1978,16:iii-xiii,1-282.
6 Jennekens FG,ten Kate LP,de Visser M,et al.Diagnostic criteria for Duchenne and Becker muscular dystrophy and myotonic dystrophy.Neuromuscul Disord,1991,1:389-391.
7 Pescatori M,Broccolini A,Minetti C,et al.Gene expression profiling in the early phases of DMD:a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression.FASEB J,2007,21:1210-1226.
8 Koenig M,Hoffman EP,Bertelson CJ,et al.Complete cloning of the Duchenne muscular dystrophy(DMD)cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals.Cell,1987,50:509-517.
9 Kunkel LM,Monaco AP,Hoffman E,et al.Molecular studies of progressive muscular dystrophy(Duchenne).Enzyme,1987,38(1-4):72-75.
10 De Pasquale L,D'Amico A,Verardo M,et al.Increased muscle expression of interleukin-17 in Duchenne muscular dystrophy.Neurology,2012,78:1309-1314.
11 Altamirano F,Lopez JR,Henriquez C,et al.Increased Resting Intracellular Calcium Modulates NF-kappaB-dependent Inducible Nitric-oxide Synthase Gene Expression in Dystrophic mdx Skeletal Myotubes.J Biol Chem,2012,287:20876-20887.
12 Chen YW,Zhao P,Borup R,et al.Expression profiling in the muscular dystrophies:identification of novel aspects of molecular pathophysiology.J Cell Biol,2000,151:1321-1336.
13 Gehrig SM,van der Poel C,Hoeflich A,et al.Therapeutic potential of PEGylated insulin-like growth factor I for skeletal muscle disease evaluated in two murine models of muscular dystrophy.Growth Horm IGF Res,2012,22:69-75.
14 Head SI,Branched fibres in old dystrophic mdx muscle are associated with mechanical weakening of the sarcolemma,abnormal Ca2+transients and a breakdown of Ca2+homeostasis during fatigue.Exp Physiol,2010,95:641-656.
15 Gorospe JR,Tharp MD,Hinckley J,et al.A role for mast cells in the progression of Duchenne muscular dystrophy?Correlations in dystrophin-deficient humans,dogs,and mice.J Neurol Sci,1994,122:44-56.
16 Irizarry RA,Hobbs B,Collin F,et al.Exploration,normalization,and summaries of high density oligonucleotide array probe level data.Biostatistics,2003,4:249-264.
17 Smyth GK,Michaud J,Scott HS.Use of within-array replicate spots for assessing differential expression in microarray experiments.Bioinformatics,2005,21:2067-2075.
18 Turner PR,Schultz R,Ganguly B,et al.Proteolysis results in altered leak channel kinetics and elevated free calcium in mdx muscle.J Membr Biol,1993,133:243-251.
19 Spencer MJ,Walsh CM,Dorshkind KA,et al.Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity.J Clin Invest,1997,99:2745-2751.
20 McDouall RM,Dunn MJ,Dubowitz V.Nature of the mononuclear infiltrate and the mechanism of muscle damage in juvenile dermatomyositis and Duchenne muscular dystrophy.J Neurol Sci,1990,99:199-217.
21 Cai B,Spencer MJ,Nakamura G,et al.Eosinophilia of dystrophin-deficient muscle is promoted by perforin-mediated cytotoxicity by T cell effectors.Am J Pathol,2000,156:1789-1796.
22 Granchelli JA,Pollina C,Hudecki MS.Duchenne-like myopathy in double-mutant mdx mice expressing exaggerated mast cell activity.J Neurol Sci,1995,131:1-7.
23 Nahirney PC,Dow PR,Ovalle WK.Quantitative morphology of mast cells in skeletal muscle of normal and genetically dystrophic mice.Anat Rec,1997,247:341-349.
24 Spencer MJ,Montecino-Rodriguez E,Dorshkind K,et al.Helper(CD4(+))and cytotoxic(CD8(+))T cells promote the pathology of dystrophin-deficient muscle.Clin Immunol,2001,98:235-243.
25 Wehling M,Spencer MJ,and Tidball JG.A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice.J Cell Biol,2001,155:123-131.
26 Serra F,Quarta M,Canato M,et al.Inflammation in muscular dystrophy and the beneficial effects of non-steroidal anti-inflammatory drugs.Muscle Nerve,2012,46:773-784.
27 Chen YW,Nagaraju K,Bakay M,et al.Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy.Neurology,2005,65:826-834.
28 Franco A,Lansman JB.Calcium entry through stretch-inactivated ion channels in mdx myotubes.Nature,1990,344:670-673.
29 Hopf FW,Turner PR,Denetclaw WF,et al.A critical evaluation of resting intracellular free calcium regulation in dystrophic mdx muscle.Am J Physiol,1996,271:C1325-1339.
30 De Backer F,Vandebrouck C,Gailly P,et al.Long-term study of Ca(2+)homeostasis and of survival in collagenase-isolated muscle fibres from normal and mdx mice.J Physiol,2002,542:855-865.
31 Niebroj-Dobosz I,Hausmanowa-Petrusewicz I.The involvement of oxidative stress in determining the severity and progress of pathological processes in dystrophin-deficient muscles.Acta Biochim Pol,2005,52:449-452.
32 Anderson JE.Arole for nitric oxide in muscle repair:nitric oxide-mediated activation of muscle satellite cells.Mol Biol Cell,2000,11:1859-1874.
33 MacLennan DH,Kranias EG,Phospholamban:a crucial regulator of cardiac contractility.Nat Rev Mol Cell Biol,2003,4:566-577.
34 Haghighi K,Gregory KN,Kranias EG.Sarcoplasmic reticulum Ca-ATPase-phospholamban interactions and dilated cardiomyopathy.Biochem Biophys Res Commun,2004,322:1214-1222.
35 Teichmann MD,Wegner FV,Fink RH,et al.Inhibitory control over Ca(2+)sparks via mechanosensitive channels is disrupted in dystrophin deficient muscle but restored by mini-dystrophin expression.PLoS One,2008,3:e3644.
猜你喜欢
科学与社会(2022年1期)2022-04-19
莫愁(2019年36期)2019-11-13
中学生数理化·高一版(2016年7期)2016-12-07
试题与研究·中考化学(2016年1期)2016-09-30
中国病理生理杂志(2015年8期)2015-12-21
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
中学科技(2015年8期)2015-08-08
医学研究杂志(2015年3期)2015-06-10
营销界(2015年22期)2015-02-28
海峡姐妹(2015年6期)2015-02-27
