
气相色谱法测定大米中马拉硫磷的不确定度评定
2015-03-29 09:09:16俞慧红
安徽农业科学 2015年35期
俞慧红
(慈溪市食品药品检验检测中心,浙江慈溪 315300)
气相色谱法测定大米中马拉硫磷的不确定度评定
俞慧红
(慈溪市食品药品检验检测中心,浙江慈溪 315300)
[目的]对气相色谱法测定大米中马拉硫磷的不确定度进行评定。[方法]建立了气相色谱法测定大米中马拉硫磷的测量不确定的数学模型,分析了不确定度的主要来源,对该方法所得结果的已识别来源的不确定度进行评价,并计算相对合成不确定度和相对扩展不确定度。[结果]该研究测定过程所产生的测量不确定度主要来源于回收率和重复性试验,其他因素影响很小,用该法测得大米中马拉硫磷含量为0.008 58 mg/kg,扩展不确定度为0.000 51 mg/kg(95%,k=2)。[结论]该评价方法及结果对提高马拉硫磷检测数据的可靠性与准确性有一定的指导意义。
气相色谱;马拉硫磷;不确定度
大米是主要的粮食之一,在人们食品消费中占有很大的比例。随着人们食品安全意识的提高,大米中农药残留越来越引起人们的关注。马拉硫磷作为一种高效低毒的有机磷杀虫剂,是常用的防治农业害虫的化学农药,国家标准GB2763-2014《食品安全标准 食品中农药最大残留限量》[1]明确规定大米中马拉硫磷的最大残留限量为0.1 mg/kg,因此,马拉硫磷检测的准确性显得至关重要。
测量不确定度是表征合理地赋予被测量之值的分散性,与测量结果相联系的参数,为一非负参数,即恒为正值[2]。测量不确定度是测量结果质量的衡量尺度,合理评定测量结果的不确定度是分析实验室必须重视的问题。根据JJF 1059.1-2012《测量不确定度评定与表示》,检测实验室对有关测量不确定度的问题进行了广泛的研究[3-9],马拉硫磷的检测方法参考GB/T 5009.20-2003食品中有机磷农药残留量的测定[10],这是测定有机磷农药残留量的常用方法之一,关于该方法的研究已屡见报道[11-13],而有关气相色谱法检测马拉硫磷含量的不确定度却鲜有报道。笔者结合GB/T 5009.20-2003检测方法和实际操作,以简化评定、不遗漏、不重复为原则,建立数学模型,分析测量结果的不确定度来源并定量,对测量结果进行不确定度评定,以期在实际检测工作中严格控制影响测量不确定度的主要因素,尽可能降低测量结果的不确定度,提高检测结果的准确性与可靠性。
1 材料与方法
1.1 材料
主要试剂:丙酮、二氯甲烷、氯化钠、无水硫酸钠、马拉硫磷标准溶液,农业部环境保护科研监测所,浓度值100 mg/L。主要仪器设备:7890A气相色谱仪(FPD, 美国安捷伦公司),色谱柱 DB1701(30 m×0.25 mm×0.25 μm),天平(0.01 g),粉碎机,匀浆机,旋转蒸发仪。
1.2 检测方法
准确称取25.00 g制备好的粉碎后大米样品于250 ml广口瓶中,加入50 ml水和100 ml丙酮,用高速匀浆机匀浆2 min,静置10 min,使丙酮和水相分层,水相再用50 ml二氯甲烷振摇2 min,静置分层,将丙酮和二氯甲烷提取液合并经装有无水硫酸钠的玻璃漏斗脱水过滤至KD浓缩瓶,再以少量二氯甲烷洗涤容器和无水硫酸钠,合并洗涤液至浓缩瓶,用旋转蒸发仪减压浓缩至近干,浓缩液用二氯甲烷转移定容至10 ml,摇匀待测,进样1 μl,外标法定量[14]。
2 数学模型
2.1 数学模型
大米中马拉硫磷含量的计算公式如下:
(1)
式中,C为样品中的马拉硫磷含量(mg/kg);C样为由标准曲线求得样液中马拉硫磷的浓度(μg/ml);V为样液最终定容体积( ml);m为称取的试样量(g);frep为重复性因子;R为方法回收率。
由公式(1)可得合成不确定度为:
Urel(C)=
(2)
2.2 不确定度分量的主要来源
该试验主要根据马拉硫磷建立的数学模型,对数学模型中各个参数包括由标准曲线求得样液中马拉硫磷含量、样品处理(定容体积与样品称重)、重复性试验以及回收率所引起的不确定度进行分析及定量。
3 结果与分析
3.1 由标准曲线求得样液中马拉硫磷含量引入的相对不确定度urel(C样)
样液马拉硫磷的浓度不确定度u(C样)包括标准使用液配制过程中的不确定度u(C标)和校正曲线拟合引起的不确定度u(curve)。
3.1.1 标准储备溶液及稀释过程中引入的相对标准不确定度urel(C标)。标准溶液配制过程:吸取1 ml马拉硫磷存储标准溶液100 μg/ ml定容至25 ml容量瓶,配成4 μg/ml中间标准溶液,再各移取1 ml中间标准溶液定容至5、10、25、50、100 ml容量瓶,配成标准使用溶液:0.04、0.08、0.16、0.40、0.80 μg/ml。
标准使用液配制过程中相对标准不确定度主要影响因素有:标准品相对不确定度u1rel(C标);配制标准使用液时所用1 ml单标移液管引入的不确定度u2rel(C标);配制标准使用液时所用5、10、25、50、100 ml A级单标容量瓶引入的相对不确定度u3rel(C标)、u4rel(C标)、u5rel(C标)、u6rel(C标)、u7rel(C标)。
标准使用液配制过程中相对不确定度urel(C标)的数学模型为:

(3)


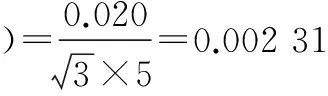

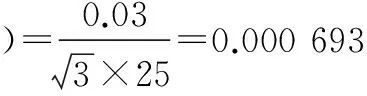
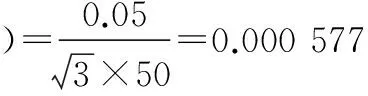
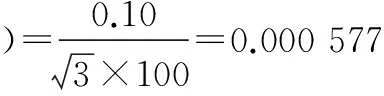
将各分量带入公式(3),标准溶液配制相对合成标准不确定度为:urel(C标)=0.004 93。
3.1.2 校正标准曲线的相对不确定度urel(curve)。校正标准曲线各点马拉硫磷的浓度和仪器响应的峰面积列于表1。校正曲线线性方程为:
Aj=aCi+b

表1 校正曲线的浓度和仪器响应的峰面积
式中,Aj为第j个校正溶液对应的第j次测定峰面积;Ci为第j个校正溶液的浓度;a为斜率,为9 663.079 62;b为截距,为-59.112 96。采用最小二乘法拟合曲线,校正曲线线性相关系数r为0.999 93。对同一个样品进行2次测量,由曲线求得马拉硫磷的含量为0.043 6 μg/ml。标准曲线的相对标准不确定度为
(4)
由于C样不确定度包括标准使用液配制过程中的不确定度u(C标)和校正曲线的不确定度u(curve),因此,由标准曲线求得的样液中马拉硫磷的浓度引入的相对不确定度为:

3.2 由样品处理引入的相对标准不确定度urel(m)和urel(V)
样品处理引入的相对不确定度主要包括称量样品所使用的天平引入的相对标准不确定度urel(m)和样品处理溶液定容时所用的10 ml容量瓶引入的相对标准不确定度urel(V)。

由于样品称量均为50.00 g,称量样品引起的相对标准不确定度为:
样品处理溶液定容时所用的10 ml容量瓶通过JJG196-2006《常用玻璃量器检定规程》查得,10 ml A级容量瓶允许最大误差为±0.020 ml,则样品定容引起的相对不确定度为:
3.3 重复性试验引入的相对标准不确定度urel(frep)

则重复性引起的相对标准不确定度为:
3.4 回收率的相对不确定度urel(R)
对空白样品进行12次加标回收试验,加标水平为0.016 mg/kg,回收率试验见表2。

表2 加标回收率试验
由表2可以知道,回收率平均值R=0.963%,标准偏差(s)=0.009 3,用t检验法来检验回收率是否与100%存在显著性差异。
f=n-1=11,查t值表得,t0.05,11=2.20,t>t0.05,11,P<0.05。因此,平均回收率R与100%存在显著性差异。方法使用有证标准物质做回收试验,其不确定度为:
式中,s为重复分析的标准偏差,s=0.009 3;R为平均回收率0.963;n为重复次数,12次;cs为标准物质证书给定的标准值。
由于标准物质的不确定度已于标准使用溶液引入的不确定度已考虑过,这里不做再次考虑,上式简化为:
由于回收率R与100%存在显著性差异,需要进行修正,修正公式如下:
3.5 合成不确定度
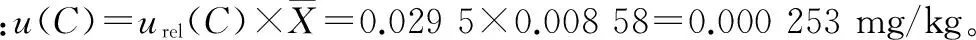
3.6 扩展不确定度
根据测量不确定度评定指南对一般实验室的要求,在置信概率P=95%时,测量结果的扩展不确定度包含因子k=2,则其扩展不确定度为:
U=2×0.000 253=0.000 51 mg/kg。马拉硫磷的测定结果为:(0.008 58±0.000 51)mg/kg,k=2。
4 讨论
通过对不确定度的计算评定,比较了各相对标准不确定度分量,影响测定大米中马拉硫磷的不确定度主要因素是回收率和重复性试验,样液浓度次之,样品处理包括定容和称重影响最小,因此,回收率与重复性试验引入的不确定度不能忽略,具体如下:由标准曲线求得样液中马拉硫磷含量,量值0.043 6 mg/L,相对标准不确定度0.006 96,贡献率14.30%;样品处理(定容),量值10 ml,相对标准不确定度0.001 15,贡献率2.40%;样品处理(称重),量值25.00 g,相对标准不确定度0.000 230 8,贡献率0.20%;重复性试验,量值0.008 58 mg/kg,相对标准不确定度0.019 0,贡献率39.00%;回收率,量值0.963%,相对标准不确定度0.021 5,贡献率44.1%。
对气相色谱法测定大米中马拉硫磷测量结果进行不确定度分析与定量,一方面能了解被检测指标真值所处范围及其大小,减少马拉硫磷的判定风险;另一方面,在实际检测工作中,控制影响测量不确定度的主要因素,对样品进行加标回收和平行试验,以回收率折算后的平均值作为检测结果,提高检测数据的可靠性和准确性。
[1] 中华人民共和国国家卫生和计划生育委员会.食品中农药最大残留限量:GB 2763-2014[S].北京:中国标准出版社,2014.
[2] 国家质量技术监督局.测量不确定度评定与表示:JJF 1059.1-2012[S].北京:中国标准出版社,2014.
[3] 姚祖江,魏超,谢君红,等.气相色谱法测定蔬菜中有机磷残留量不确定度评定[J].现代农业科技,2015(19):143-146.
[4] 李芳,王兴磊,尚爽,等.气相色谱法测定果蔬中毒死蜱的不确定度评定[J].广州化工,2014(22):136-138,180.
[5] 殷朝珍,翟付凤,董卫峰.气相色谱法测定蔬菜中有机磷农药残留量不确定度评定研究[J].现代农业科技,2014(17):153-155.
[6] 薛丽,孙茜,刘敏,等.气相色谱法测定蔬菜水果中有机氯和拟除虫菊酯类农药残留量的不确定度评定[J].粮油加工(电子版),2015(1):60-62,65.
[7] 黄永忠.气相色谱法测定白酒中乙酸乙酯的不确定度评定[J].广州化工,2015(2):90-92,132.
[8] 刘佳,王俊澄,张辉.气相色谱法测定熟肉制品中甜蜜素含量的不确定度评定[J].轻工科技,2014(6):15-16.
[9] 曹小彦,张燕,黄雄伟,等.气相色谱法测定葡萄酒中甲醇含量不确定度评定[J].食品与机械,2013(6):76-78,175.
[10] 中华人民共和国国家卫生部中国国家标准化管理委员会.食品中有机磷农药残留量的测定:GB/T 5009.20-2003[S].北京:中国标准出版社,2004.
[11] 王江蓉.稻谷中马拉硫磷残留气相色谱分析方法研究[J].粮食加工,2008(4):76-77.
[12] 耿玉辉,郭桂金.气相色谱法测定粮食中农药残留——马拉硫磷[J].粮食与食品工业,2007(2):50-51.
[13] 肖青,陈树.气相色谱法测定大米中马拉硫磷的残留[J].现代食品科技,2007(12):83-84.
Evaluation of Uncertainty for Determination of Malathion in Rice with GC
YU Hui-hong
(Cixi food and drug inspection and Testing Center, Cixi, Zhejiang 315300)
[Objective] The study was to evaluate the uncertainty of measuring malathion residue in rice with GC.[Method] The establishment of mathematical model of the measurement of malathion in rice by GC was uncertain, the main sources of uncertainty were analyzed, the influence of the identified uncertainty of the analytical result was evaluated.And the relative synthesis standard uncertainty and the relative expansion uncertainty were calculated out.[Result] The measurement uncertainty of the measurement process was mainly derived from the recovery rate and repeatability experiments, other factors were very small.The method for measuring the content of rice malathion was 0.008 58 mg/kg,the expanded uncertainty was 0.000 51 mg/kg (95%,k=2). [Conclusion] The evaluation method and results have certain guiding significance to improve the reliability and accuracy of the detection data of malathion.
GC;Malathion;Uncertainty
俞慧红(1979- ),女,浙江新昌人,工程师,从事食品质量管理和检测工作。
2015-11-04
TS 212.7
A
0517-6611(2015)35-148-03
猜你喜欢
工业用水与废水(2022年3期)2022-06-29 04:04:22
昆钢科技(2021年2期)2021-07-22 07:46:56
中国兽药杂志(2021年1期)2021-04-19 11:33:06
色谱(2019年4期)2019-04-02 12:10:12
职工法律天地(2018年12期)2018-01-22 22:55:10
水利信息化(2017年4期)2017-09-15 12:01:21
畜牧兽医科技信息(2016年8期)2016-02-22 01:14:12
电源技术(2015年7期)2015-08-22 08:48:52
电测与仪表(2015年7期)2015-04-09 11:40:30
电测与仪表(2014年9期)2014-04-15 00:27:16
