
原位聚合法制备碳十八甲基丙烯酸酯聚合物及其用作开管毛细管电色谱固定相
2015-03-24 06:40周孙英陈继涢谭静静林旭聪谢增鸿
色谱 2015年12期
周孙英, 陈继涢, 谭静静, 林旭聪, 谢增鸿
(1. 福建医科大学药学院, 福建 福州 350108; 2. 福州大学食品安全和环境监测研究所, 福建 福州 350002)
研究论文
原位聚合法制备碳十八甲基丙烯酸酯聚合物及其用作开管毛细管电色谱固定相
周孙英1*, 陈继涢1, 谭静静2, 林旭聪2, 谢增鸿2
(1. 福建医科大学药学院, 福建 福州 350108; 2. 福州大学食品安全和环境监测研究所, 福建 福州 350002)
以十八碳醇甲基丙烯酸酯为单体、乙二醇二甲基丙烯酸酯为交联剂,采用原位聚合法合成了一种新型毛细管开管柱固定相,优化了毛细管开管柱的制备参数。柱内表面的电镜图像显示其具有多孔皱褶、质地均匀的结构特征。将其应用于甲苯、乙苯、丙苯、丁苯、戊苯和己苯的分离试验中,6种化合物达到了完全分离,出峰顺序与它们的疏水性一致,表明该柱有明显的疏水色谱作用。在10 mmol/L磷酸盐(pH 8.5,含50%(v/v)乙腈)流动相、16 kV电压下,该开管柱成功地分离了4种抗癫痫类药物,柱效范围为35 300~49 800 塔板/m,与空柱管相比分离效果明显提高。结果表明通过本实验的原位聚合法可制备具有反相色谱作用的有机基质碳十八开管毛细管电色谱柱。
原位聚合;固定相;十八碳醇甲基丙烯酸酯;开管毛细管电色谱;抗癫痫药
开管柱制备过程简单、易于控制,柱效高,且开管柱毛细管电色谱(OT-CEC)中的电渗流(EOF)比填充毛细管电色谱(P-CEC)中的EOF高60%[1],更适用于快速分析。但是它最大的缺点是相比小,对溶质的保留有限、柱容量低。因此,如何制备高表面积、高配基容量、高效率的开管毛细管电色谱柱是研究的热点。
目前,提高OT-CEC固定相表面积的制柱方法有蚀刻法(表面粗糙化后键合固定相,其制柱过程是先对其表面进行蚀刻,以增大表面积,然后将固定相键合上去)[2]、溶胶-凝胶(sol-gel)法[3,4]、多孔聚合物涂层法[5,6]和纳米粒子涂层法[7,8]。由于纳米材料制备困难,其应用受到限制,所以较常用的是前3种方法。蚀刻法使用NH4HF2为毛细管表面腐蚀剂,其腐蚀性强,操作时需采取人体保护措施,而且制柱操作较繁琐,周期长。对sol-gel法来说,凝胶的形成和其微观结构与溶胶的配制密切相关,需要按照严格的成分比例配制溶胶,形成的溶胶稠度与温度和时间等直接相关,所以要制备重现性好的sol-gel是一项很难掌握的技术,也影响了柱的重现性。原位聚合法将预聚合溶液注入毛细管后,一体化形成聚合物涂层,预聚合溶液的配制与整体柱相似,操作较容易,重现性好。
为了提高聚合物中配基的容量,有效地改善分离性能,本实验选择分子较大的具有十八烷基的正十八碳醇甲基丙烯酸酯作为色谱功能单体,由此制备具有反相色谱作用的开管柱固定相。类似的反相色谱开管柱,曾有报道用sol-gel技术制作C18[9,10]、C8[11]开管柱固定相。本实验采用原位聚合法制备有机基质的C18开管电色谱固定相,提供了一种新型毛细管开管柱。
1 实验部分
1.1 仪器与试剂
开管毛细管电色谱仪由实验室自组装:高压电源(±30 kV/0.3 mA,中国科学院上海物理研究所);1010-LC高效液相色谱泵、紫外-可见波长柱上检测器(Trisep®-2100, Unimicro Technologies, Pleasanton, CA, USA); N2000数据采集软件(南京千谱软件公司); Philips XL30扫描电子显微镜(Philips, Eindhoven, 荷兰); PHS-3C型精密酸度计(上海大普仪器有限公司); DKB-501A型超级恒温水槽(上海精宏实验设备有限公司); 85-1型恒温磁力搅拌器(江苏金坛国华仪器公司); GC-4000A Series气相色谱柱温箱(北京东西电子研究所); KQ-100B型超声清洗器(昆山市超声仪器有限公司)。
正十八碳醇甲基丙烯酸酯(octadecyl methacrylate, OMA)、乙二醇二甲基丙烯酸酯(ethylene dimethacrylate, EDMA)、3-(甲基丙烯酰氧)丙基三甲氧基硅烷(3-(trimethoxysilyl)propyl methacrylate,γ-MAPS)、甲基丙烯酸(methacrylic acid, MAA)(Aldrich, Milwaukee, WI, USA);乙苯、丙苯、丁苯、戊苯和己苯(Sigma, St. Louis, MO, USA);偶氮二异丁腈(2,2-azobisisobutyronitrile, AIBN)(分析纯,上海试剂四厂);甲苯、硫脲、N,N-二甲基甲酰胺(N,N-dimethylformamide, DMF)、磷酸(分析纯,中国医药集团上海化学试剂公司);苯巴比妥、卡马西平、异戊巴比妥(分析纯,中国医药集团上海化学试剂公司)、苯妥英钠(东京化成工业株式会社);乙腈(色谱纯,中国医药集团上海化学试剂公司);实验用水由Milli-Q超纯水系统提供;熔融石英毛细管(内径50 μm,外径375 μm,河北永年锐泽色谱器件有限公司)。
1.2 电色谱实验
流动相的配制:缓冲溶液经0.22 μm微膜过滤,将适量体积的乙腈、缓冲溶液混合,运行前超声20 min脱气。实验前用缓冲溶液冲洗毛细管柱15 min后,在电色谱仪器上施加电压平衡至获得稳定的电流和基线信号。每次更换流动相时需用新的流动相平衡15 min。柱温保持室温(25 ℃)。采用电动方式进样(10 kV、3 s)。硫脲为电渗流标记物。
1.3 开管柱的制备
取一定长度的经过预处理后的毛细管,将30%的硅烷化试剂γ-MAPS甲苯(无水)溶液注满毛细管,密封,加热至110 ℃反应12 h。用甲醇洗涤30 min, 通氮气60 ℃下吹干。
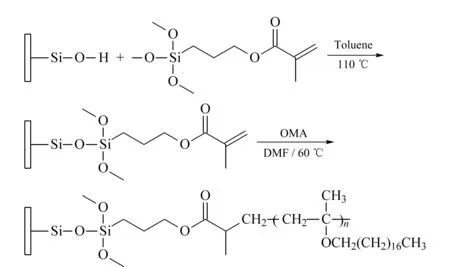
图 1 毛细管内壁修饰聚合物的反应示意图Fig. 1 Reaction scheme for the modification of the capillary inner wall with grafted polymer chains
聚合反应液的质量为2.0 g,单体与致孔剂的质量比为25∶75。单体体系成分包括:色谱功能单体OMA 84%(质量分数,相对于单体总量,下同)、电渗流产生试剂MAA 5%、交联剂EDMA 10%、引发剂AIBN 1%。单体体系溶解于致孔剂DMF中。然后超声反应混合液,使其充分溶解澄清,继续用氮气吹10 min。将一小部分反应混合物吸入已经硅烷化过的长度为65 cm的空毛细管中,停留20 min,将毛细管的两端用橡胶小塞封口,并浸于60 ℃恒温水浴箱内,反应6 h后用氮气吹毛细管,除去未固化的材料,在管壁上留下一层涂层,继续将毛细管的两端用橡胶小塞封口,浸于60 ℃恒温水浴箱内24 h。反应原理如图1所示。反应完成后,用甲醇冲洗毛细管1 h。在毛细管离出口端约20 cm处烧一个1~2 mm的检测窗口,并在放大镜或显微镜下确认内部涂层未被破坏。切一段2 cm的毛细管柱用于电镜分析。
2 结果与讨论
2.1 聚合反应条件的优化
2.1.1 单体浓度的优化
聚合反应液中,单体浓度直接影响固定相中色谱配基的密度和固定相的微观结构及对溶质的保留,体现在对分离效率的影响。保持交联剂和引发剂比例不变,改变单体体系浓度(10%~30%(质量分数)),以硫脲和甲苯为对象,考察开管柱对两者的分离情况(见图2)。从图2中看到,单体浓度从10%逐渐增大到25%,硫脲与甲苯的分离度也随之提高,甲苯的保留时间增加,反映了固定相疏水作用增强,而且峰形也随之变窄,即效率逐渐升高。所以随着单体浓度的增大,其在固定相中的密度增大,当单体浓度增加至25%后接近饱和(见图2),单体浓度继续增大至30%,其分离结果与25%时基本相同。因此单体在聚合反应液中适宜的浓度为25%。
2.1.2 聚合溶液中MAA含量的选择
表1中列出的实验结果是采用不同含量的MAA单体体系制备的毛细管柱,以硫脲作为电渗流标记物的迁移时间和相应的电渗流大小。从表1看到,采用不含MAA的单体体系制备毛细管柱,硫脲在18 min之后出峰,EOF较小。为了减少分析时间,提高效率,在固定相中加入适量的可电离为负离子基团的物质(MAA)。单体体系中MAA含量从3%增至8%,硫脲的迁移时间随之缩短,其中MAA含量从5%增至8%时硫脲的迁移时间缩短约1 min,变化渐缓,因此本文选择单体体系中MAA的含量为5%。

图 2 不同单体浓度(10%~30%)制得的C18-OT柱分离硫脲和甲苯的电色谱图Fig. 2 Electrochromatograms of the separation of toluene and thiourea on C18-OT columns prepared with different monomer concentrations (10%-30%) Conditions: capillary, C18-OT, 60.0 cm total length (40.0 cm effective length)×50 μm i.d; mobile phase, 70%(v/v) acetonitrile in 10 mmol/L phosphate buffer (pH 7.0); applied voltage, 16 kV; detection wavelength, 214 nm. Peaks: 1. thiourea; 2. toluene.表 1 单体体系中MAA含量对毛细管柱电渗流的影响Table 1 Effect of MAA content in the monomer mixture on the EOF of the resulting columns
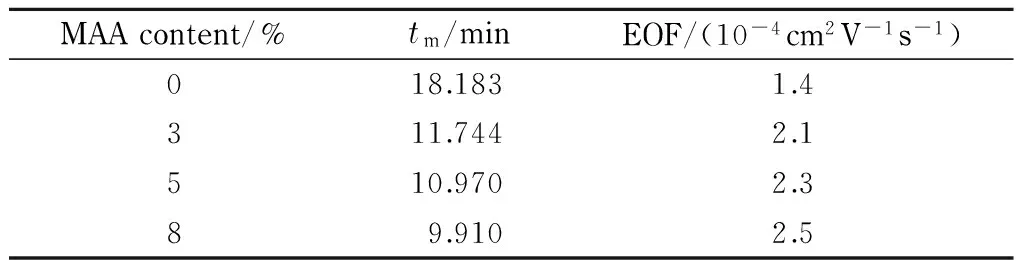
MAAcontent/%tm/minEOF/(10-4cm2V-1s-1)018.1831.4311.7442.1510.9702.389.9102.5
The conditions are the same as in Fig. 2.tm: migration time.
2.1.3 溶剂的选择
分别以甲苯、环己醇和DMF为溶剂制备毛细管柱。3种试剂中,单体在甲苯中的溶解性最好,但毛细管柱易堵塞,反应进行到3.5 h之后,毛细管柱完全堵塞。这可能是因为甲苯为非极性,单体在其中溶解性好,分子链更舒展,反应易进行;再有聚合物在甲苯中的溶解性也较好,其聚合度高,因此毛细管柱易堵塞,制柱较困难。以环己醇和DMF为溶剂,反应进行到6 h,聚合体系依然为液态,通氮气即可吹出。溶剂的极性大,聚合物在其中的溶解性小,则易沉积下来,故采用具有一定极性的试剂,既可以作为溶剂也可以作为致孔剂。采用不同极性的溶剂制备的固定相的结构不同。图3为开管柱的横截面扫描电镜图。图3a和3b为在DMF溶剂中所制备的开管柱的不同放大倍数的形貌,从图3中可看出管壁均匀地修饰上了聚合物涂层,具有明显的皱褶,而且涂层的结构均匀有序,涂层与毛细管管壁结合得很好,没有脱落和缝隙,与图3d中裸柱光滑的内表面相比较,此质地的涂层具有较大的比表面积,相对较大的柱容量。图3c为在环己醇溶剂中制备的毛细管柱,涂层具有不同的纹理结构。
采用不同极性的溶剂制备的固定相的表面结构有差别,柱性能也有差异。图4a和图4b分别为以环己醇和DMF为溶剂制备的毛细管柱分离6种烷基苯的电色谱图。比较两张色谱图可见图4b的分离效率明显地高于图4a。因此本文选择DMF为溶剂制备毛细管柱。

图 3 C18-OT柱横截面扫描电镜图Fig. 3 Scanning electron microscope (SEM) photographs of inner surface of C18-OT columnsa and b. 5 000× and 20 000× (column prepared in N,N-dimethylformamide); c. 5 000× (column prepared in cyclohexane); d. the bare capillary.

图 4 烷基苯同系物的电色谱分离图Fig. 4 Electrochromatograms of alkyl benzene homologous series on C18-OT columns prepared in (a) cyclohexane and (b) DMF solvents Conditions: capillary, C18-OT, 58.0 cm total length (38.0 cm effective length)×50 μm i.d.; mobile phase, 60%(v/v) acetonitrile in 10 mmol/L phosphate buffer (pH 7.0); applied voltage, +16 kV; detection wavelength, 214 nm. Peaks: 0. thiourea; 1. toluene; 2. ethylbenzene; 3. propylbenzene; 4. butylbenzene; 5. amylbenzene; 6. hexylbenzene.
2.2 毛细管柱的电色谱性能表征
2.2.1 pH值对电渗流的影响
图5中曲线b为流动相的pH值对C18-OT柱电渗流的影响。pH≤7时,电渗流随pH值的升高快速上升,这是因为残余硅羟基和羧基的解离程度随pH增大而增大。但pH>7后,羧基解离趋于平衡,所以EOF的曲线变平缓。图5中曲线a为空管的电渗流与pH值的关系,其EOF随pH增大而快速上升。可见C18-OT柱表面的羧基是电渗流的主要贡献者。
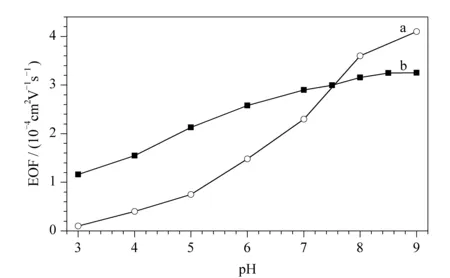
图 5 流动相中pH值对(a)空毛细管柱和(b)C18-OT柱电渗流的影响Fig. 5 Influence of pH value on EOF of (a) the bare capillary and (b) C18-OT column Conditions: capillary, 60.0 cm total length (40.0 cm effective length)×50 μm i.d.; mobile phase, 50 mmol/L citrate solution for pH 3.0-5.0 and 50 mmol/L phosphate solution for pH 6.0-9.0; applied voltage, 10 kV; detection wavelength, 214 nm.
2.2.2 C18-OT柱对烷基苯的分离
对于中性物质,其电泳淌度为0,中性化合物在电色谱中的分离主要基于溶质和固定相之间的色谱作用。在本实验中,所制备的C18-OT柱由于含有十八碳烷基疏水性基团,是典型的疏水性固定相。因此,选取疏水性不同的苯类衍生物,考察该毛细管柱的反相色谱性能。6种烷基苯(甲苯、乙苯、丙苯、丁苯、戊苯和己苯)在结构上依次只相差一个亚甲基,其疏水性随着亚甲基的增加而逐渐增强。如图6所示,分离对象按其疏水性从弱到强的顺序依次被洗脱下来:硫脲(作为不保留标记物)、甲苯、乙苯、丙苯、丁苯、戊苯和己苯,并达到了基线分离。
考察了流动相中乙腈含量(体积分数)分别为70%、60%、50%时烷基苯同系物在C18-OT柱上的分离效果。随着流动相中乙腈含量的减少,其疏水性洗脱能力减弱,疏水性溶质在固定相的保留时间延长,溶质间的分离度增大。乙腈含量为70%时甲苯和乙苯没有达到完全基线分离(见图6a)。乙腈含量为60%时,两者完全分离,其他组分的保留时间也延长(见图4b)。乙腈含量为50%时各物质之间的分离度进一步增大,但最后两个物质的出峰时间过长,峰形展宽严重(见图6b)。这些实验结果充分验证了该开管柱良好的反相色谱分离能力。
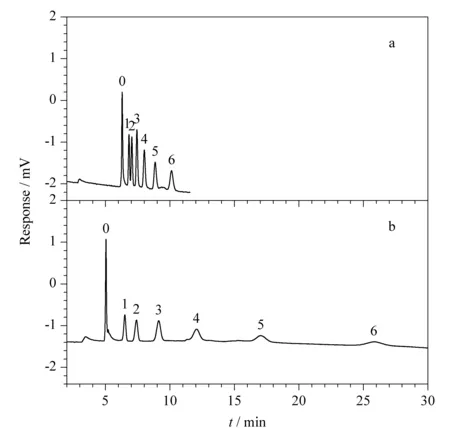
图 6 流动相中乙腈含量不同时烷基苯同系物的电色谱分离图Fig. 6 Electrochromatograms of alkyl benzene homologous series by different contents of acetonitrile in mobile phase Conditions: capillary, C18-OT, 58.0 cm total length (38.0 cm effective length)×50 μm i.d.; mobile phase, (a) 70% and (b) 50%(v/v) acetonitrile in 10 mmol/L phosphate buffer (pH 7.0); applied voltage, +16 kV; detection wavelength, 214 nm. Peaks: 0. thiourea; 1. toluene; 2. ethylbenzene; 3. propylbenzene; 4. butylbenzene; 5. amylbenzene; 6. hexylbenzene.
2.2.3 柱的重现性
在缓冲溶液为10 mmol/L磷酸盐(pH 7.0,乙腈含量50%)、电压为16 kV条件下,通过硫脲迁移时间的相对标准偏差考察了开管柱的重现性。其电渗流的日内RSD(n=5)为0.85%,日间RSD(n=5)为1.65%。该结果说明C18-OT柱的重现性良好。保持配方一样,采用不同批次的聚合溶液制备了3根开管柱。柱与柱之间电渗流的RSD(n=3)为5.8%,表明制备柱的方法重现性较好。此外,经过100次分离后,柱效并没有明显下降,说明该开管柱有较长的使用寿命。
2.3 抗癫痫药物的分析
卡马西平、苯妥英钠、苯巴比妥类是临床上常用的抗癫痫药物,在控制患者病情时,常将上述药物联合使用。但当血药浓度超过安全范围可产生毒性反应,因此分析这几种药物具有重要意义。常用的分析方法有HPLC[12,13]、胶束毛细管电泳(MECC)[14,15]。目前,用毛细管区带电泳(CZE)同时分离检测这3类物质的报道还很少。本文考察了卡马西平、苯妥英钠、苯巴比妥、异戊巴比妥在C18-OT柱上的电色谱分离情况。
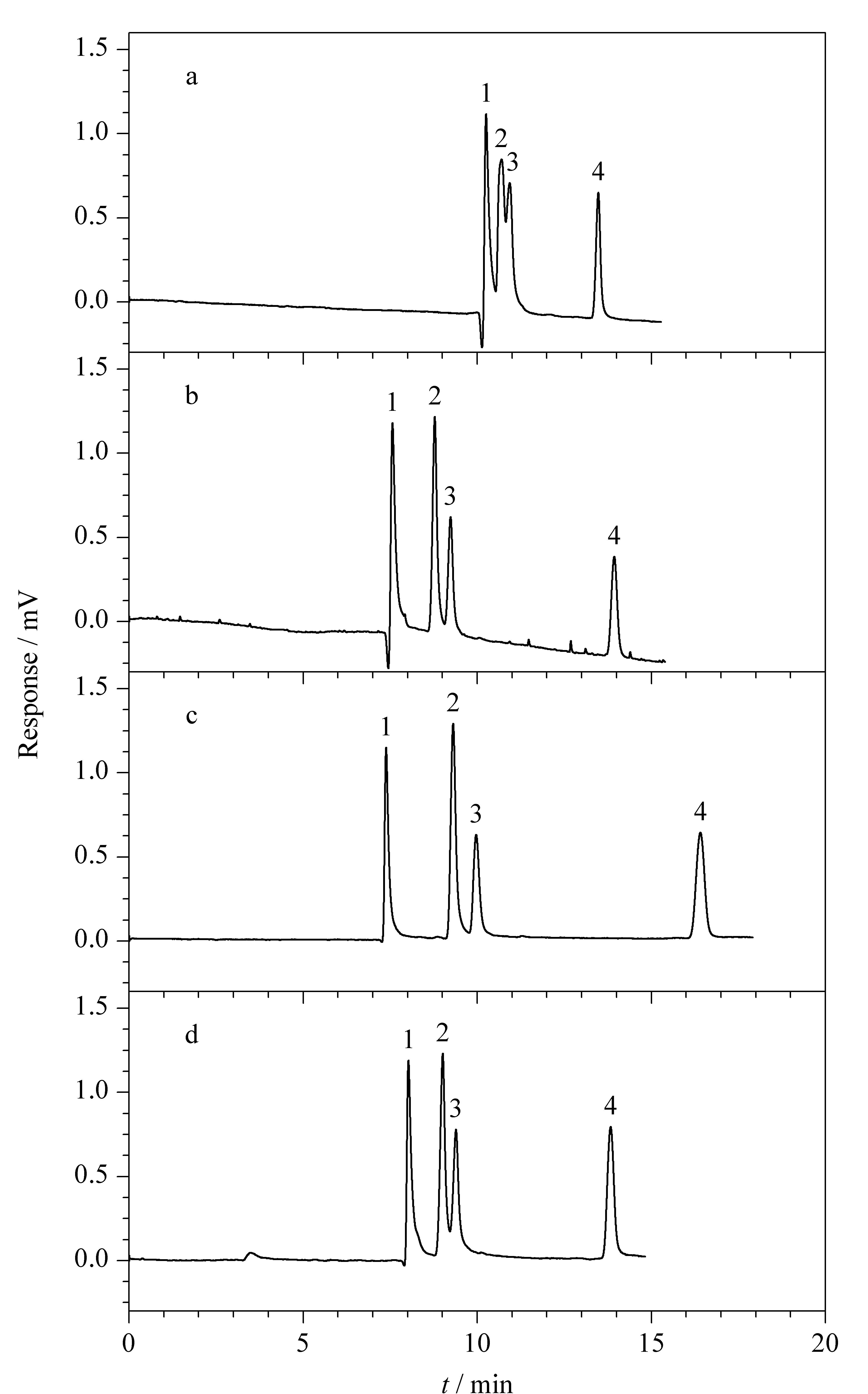
图 7 4种药物在不同pH条件下的电色谱分离图Fig. 7 Electrochromatograms of four antiepileptics at different pH values Conditions: capillary, C18-OT, 60.0 cm total length (40.0 cm effective length)×50 μm i.d.; mobile phase, 50%(v/v) acetonitrile in 10 mmol/L phosphate buffer at (a) pH 7.5, (b) pH 8.0, (c) pH 8.5 and (d) pH 9.0; applied voltage, +16 kV; detection wavelength, 214 nm. Peaks: 1. carbamazepine; 2. phenytion; 3. amobarbital; 4. phenobarbital.
2.3.1 pH值的影响
对于带电物质来说,pH值决定其带电状态,影响其电泳速率,也影响溶质的保留行为。卡马西平为电中性物质,苯妥英钠、异戊巴比妥、苯巴比妥的pKa分别为8.4、7.9、7.4。改变流动相的pH值(7.5~9.0)来考察pH值对4种药物的电色谱分离影响(见图7)。在pH为7.5时,苯巴比妥发生电离,带负电荷,其电泳方向与电渗流方向相反,导致保留时间长;苯妥英、异戊巴比妥分子只部分电离而带少量负电荷,所以出峰早于苯巴比妥,但两者荷质比相差不大,因此只达到了部分分离;卡马西平为电中性,故最早出峰。pH值升高至8.0, 3种带电化合物的电离程度增大,荷质比相差增加,分离度增大。pH为8.5时已经超过它们的pKa, 3种化合物都以负电荷存在,因此达到了完全分离。pH为9.0时,苯妥英、异戊巴比妥带有更多负电荷,两者的荷质比差别反而减小,因此分离度降低。由此可见电迁移对这4种药物的分离起重要作用。考虑到分离度和柱效,合适的流动相pH值为8.5。
2.3.2 乙腈含量和缓冲盐浓度的影响
流动相中有机试剂(乙腈)的含量影响毛细管柱的EOF,也影响流动相的洗脱能力。本实验考察了4种药物在乙腈含量为70%~30%(v/v)条件下的电色谱分离情况(图略)。乙腈含量低于40%时,各组分峰出现拖尾,这是因为乙腈含量的减少使流动相对弱极性物质的洗脱能力下降。乙腈含量高于60%时,流动相对溶质的洗脱能力增强,一方面苯妥英和异戊巴比妥的分离度随之减小,另一方面最后的组分出峰时间延长。乙腈含量增至70%时,出峰时间延至26 min,而且峰形明显展宽,这是由于乙腈含量增加,电渗流变小,负电荷质比最大的苯巴比妥反向电泳突出。乙腈含量为50%时,4种物质达到最好的分离效果。
盐浓度的大小会影响Zeta电势、双电层厚度和柱表面的电荷数等,影响EOF,进而影响溶质的分离。本实验在pH 8.5、乙腈含量为50%(v/v)条件下,考察了流动相中盐浓度从5~25 mmol/L对溶质分离的影响。结果表明,在5 mmol/L盐浓度体系中苯妥英和异戊巴比妥不能分离;当盐浓度达到10 mmol/L时能够完全分离,并且峰形对称。继续提高盐浓度,峰出现拖尾,浓度越大,拖尾越明显。这是因为盐浓度增大,流动相中离子浓度增强,极性增强,对疏水性物质的洗脱能力减弱,溶质与固定相之间的作用增强,尤其是对最迟出峰的苯巴比妥影响明显。当盐浓度大于20 mmol/L时,苯巴比妥在60 min以后出峰,相应的峰也出现更明显的拖尾。
从以上实验结果分析来看,上述4种药物在C18-OT柱上的分离机制为电迁移和色谱两种因素共存。优化的分离条件为:以10 mmol/L磷酸盐(pH 8.5,含50%乙腈)为流动相,分离电压为16 kV。优化条件下4种药物的分离谱图见图7c,对卡马西平、苯妥英、异戊巴比妥、苯巴比妥的分离柱效分别为35 300、43 400、38 400、49 800 塔板/m。在相同条件下,用空毛细管柱分离4种药物(见图8),可见C18-OT柱的分离效果较之明显提高。本实验制备的C18-OT柱为上述抗癫痫药物的分析提供了一种新的选择。

图 8 空毛细管柱分离4种药物的电色谱图Fig. 8 Electrochromatogram of the four antiepileptics on the bare capillary The conditions are the same as in Fig. 7c.The peaks are the same as in Fig. 7.
3 结论
采用一步原位聚合的方法制备了一种具有十八碳烷基的新型毛细管电色谱开管柱。该柱固定相表面为明显皱褶、质地均匀、多孔层结构,此结构具有较大的比表面积,因此提高了相比率。在C18-OT柱上对中性物质烷基苯同系物的电色谱行为进行了考察,并对可能的保留机理进行了讨论,验证了该整体柱具有良好的反相色谱分离能力。同时应用于卡马西平、苯妥英钠、异戊巴比妥和苯巴比妥4种抗癫痫类药物的电色谱分离,柱效范围为35 300~49 800 塔板/m,效果良好,该柱还有望用于其他非极性类物质的分离,为开管电色谱的应用拓展提供了一种新途径。
[1] Ettre L S. Chromatographia, 2000, 51(1/2): 7
[2] Maria T, Matyska J, Pesek J. J Chromatogr A, 2005, 1079(1/2): 366
[3] Lü H X, Li Q Y, Yu X W, et al. Electrophoresis, 2013, 34(13): 1895
[4] Rezanka P, Ehala S, Koktan J, et al. J Sep Sci, 2011, 35(1): 73
[5] Li L M, Yang F, Wang H F, et al. J Chromatogr A, 2013, 1316: 97
[6] Aydogan C. J Chromatogr B, 2015, 976/977: 27
[7] Qin S S, Zhou C R, Zhu Y X, et al. Chinese Journal of Chromatography (覃飒飒, 周超然, 朱亚仙, 等. 色谱), 2011, 29(9): 942
[8] Zhang H H, Chen J Y, Zhou S Y. Chemical Journal of Chinese Universities (张欢欢, 陈继涢, 周孙英. 高等学校化学学报), 2015, 36(4): 631
[9] Villanueva E O, Benavente F, Giménez E, et al. Anal Chim Acta, 2014, 846: 51
[10] Guihen E, Glennon J D. J Chromatogr A, 2004, 1044(1/2): 67
[11] Miller M D, Baker G L, Bruening M L, et al. J Chromatogr A, 2004, 1044(1/2): 323
[12] Baldelli S, Cattaneo D, Giodini L, et al. Clin Chem Lab Med, 2014, 53: 435
[13] Zhang X Y, Cai X X, Zhang X Y. Chinese Journal Chtomatography (张秀尧, 蔡欣欣, 张晓艺. 色谱), 2010, 28(1): 23
[14] Lin Y Y, Wang C C, Hong Y H, et al. Anal Bioanal Chem, 2013, 405(1): 259
[15] Sieradzka E, Witt K, Milnerowicz H. Biomed Chromatogr, 2014, 28(11): 1507
Preparation of octadecyl methacrylate-based polymer stationary phase by in-situ polymerization for open tubular capillary electrochromatography
ZHOU Sunying1*, CHEN Jinyun1, TAN Jingjing2, LIN Xucong2, XIE Zenghong2
(1.SchoolofPharmacy,FujianMedicalUniversity,Fuzhou350108,China;2.InstituteofFoodSafetyandEnvironmentalMonitoring,FuzhouUniversity,Fuzhou350002,China)
The preparation of porous polymethacrylate-based open tubular capilary columns by in-situ copolymerization of octadecyl methacrylate (OMA), and ethylene dimethacrylate (EDMA) inN,N-dimethylformamide (DMF) solvent are proposed. The parameters of the preparation procedure of the stationary phase are discussed in detail. The surface of the cross-section of an open tubular (OT) column by scanning electron microscope (SEM) showed wrinkle configuration, which is expedient to enhance proportion of stationary phase and column capacity. Efficient separation of six alkyl benzene homologous series (namely, toluene, ethylbenzene, propylbenzene, butylbenzene, amylbenzene and hexylbenzene) were performed by CEC using the prepared column, and their elution order was in agreement with their hydrophobicity. So, the OT columns bonded with octadecyl ligands yielded reversed-phase retention behavior toward nonpolar solutes. In addition, the separation of four antiepileptics was also investigated with satisfactory effectiveness. The column efficiency range was 35 300-49 800 plates/m. The results showed that the C18-OT column of organic matrix for reversed-phase chromatography was successfully prepared via in-situ polymerization in this work. Hereby, this new column thus offers a promising new alternative in OT-CEC and will be useful in separation science.
in-situ polymerization; stationary phase; octadecyl methacrylate (OMA); open tubular capillary electrochromatography (OT-CEC); antiepileptics
10.3724/SP.J.1123.2015.07010
福建省自然科学基金项目(2013J01037).
2015-07-09
O658
A
1000-8713(2015)12-1307-07
* 通讯联系人.E-mail:zhsy1965@sina.com.
猜你喜欢
中国典型病例大全(2022年12期)2022-05-13
化工管理(2021年7期)2021-05-13
科学与财富(2021年33期)2021-05-10
农药科学与管理(2019年8期)2019-11-23
绿色科技(2017年16期)2017-09-22
海峡科技与产业(2017年3期)2017-04-13
红外技术(2017年1期)2017-03-27
国际检验医学杂志(2016年20期)2016-11-19
淮海医药(2015年2期)2016-01-12
中西医结合心脑血管病杂志(2014年10期)2014-05-29
