
系列Ru(Ⅱ)配合物的DNA光裂解及光谱性质的理论研究
2015-03-23 05:04王娜丽王晴晴苗体方孙国栋宋忠奥
原子与分子物理学报 2015年5期
王娜丽, 王晴晴, 苗体方,2, 孙国栋, 宋忠奥, 石 杰
(1. 淮北师范大学化学与材料科学学院, 淮北 235000; 2. 理论化学计算国家重点实验室, 长春 130023)
系列Ru(Ⅱ)配合物的DNA光裂解及光谱性质的理论研究
王娜丽1, 王晴晴1, 苗体方1,2, 孙国栋1, 宋忠奥1, 石 杰1
(1. 淮北师范大学化学与材料科学学院, 淮北 235000; 2. 理论化学计算国家重点实验室, 长春 130023)
用密度泛函理论,对系列钌多吡啶配合物1-3的电子结构、DNA光裂解及光谱性质进行了研究.首先,计算了配合物1-3的氧化还原电势,根据配合物1-3激发态还原电势的大小,合理地解释了配合物1-3的DNA光裂解能力.其次,根据配合物1-3的电子结构性质,设计了具有较高激发态还原电势的配合物4,从理论上预测配合物4具有较强的光裂解能力.最后,用TDDFT方法,在水溶液中对配合物1-3的电子吸收光谱进行了计算和模拟,计算得到的电子吸收光谱和实验结果吻合较好,实验上测得的较强吸收带从理论上被详细地解释,并研究了配合物的主配体对电子吸收光谱性质的影响.
Ru(Ⅱ)配合物; 密度泛函方法; DNA光裂解; 光谱性质
1 引 言
Ru(Ⅱ)多吡啶配合物因在光化学、光物理和生物化学中的广泛应用引起了人们极大的关注.这类以金属离子为中心的八面体钌多吡啶配合物被认为是一类较好的DNA识别试剂,在DNA结构探针、DNA光裂解、DNA分子光开关[1]、DNA介导的电子转移、抗癌药物、非线性光学材料以及生物传感器等方面应用广泛.近年来,尤其是钌多吡啶配合物的DNA光裂解性质受到极大的关注,因为在光的照射下,一些钌多吡啶配合物的还原电势明显升高,能有效地裂解DNA[2-4].为了寻找高效DNA光裂解试剂,大量的钌多吡啶配合物被合成并修饰,如增加配体共轭面积,在配体上增加吸电子或推电子基团等.尽管实验上对大量钌多吡啶配合物DNA光裂解性质进行了测定和分析,但钌多吡啶配合物的电子结构性质,尤其是多吡啶配合物的电子结构和其氧化还原电势的关系还不清晰,有关钌多吡啶配合物DNA光裂解及光谱性质的理论研究还很有限,因此开展钌多吡啶配合物DNA光裂解及光谱性质还是一项很有意义的工作.
本文对系列钌配合物[Ru(bpy)2(L)]2+1-3进行理论计算,其中bpy(2, 2-联吡啶)为辅助配体,L为主配体,L= ddt (1), dta (2), dpt(3),配合物1-3的结构示意图见图1,根据配合物1-3的结构性质设计了配合物4 [Ru(bpy)2(dph)]2+(见图1).希望这些研究结果对高效DNA光裂解试剂的设计与寻找提供理论依据.
2 计算方法
采用密度泛函理论(DFT),非限制性的B3LYP方法[5,6],混合基组,即Lanl2DZ基组[7,8]计算钌原子,6-31g(d)基组计算其他的非金属原子,对基态配合物1-4进行几何构型全优化.为了证实所优化的几何构型是最稳定的,同时进行了频率计算,无虚频出现则表明得到的构型是最稳定的.


方案1 计算水溶液中氧化还原电势的热力学循环
(1)
其中,

(2)

(3)

(4)

(5)
ΔGo(1)=Go[Ru(bpy)2L]n+(aq)-
Go[Ru(bpy)2L]n+(gas)
(6)
ΔGo(2)=Go[Ru(bpy)2L](n+1)+(aq)-
Go[Ru(bpy)2L](n+1)+(gas)
(7)
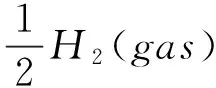
(8)
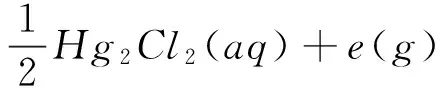
(9)
所有相关的自由能变值是在标准条件下计算得到:温度为298.15K,压力为1个标准大气压,溶液为1mol/L(水为溶剂);气体为298.15K,1个标准大气压.
电子吸收光谱的计算,以优化的基态几何结构为基础,在B3LYP/LanL2DZ水平上采用TDDFT方法在水溶液中计算200个单重激发态,根据计算的结果模拟出电子吸收光谱图.文章中所有涉及到溶液中的计算,都是以水作为溶剂,采用连续极性导体模型CPCM方法[12,13]进行计算,所有计算都是用Gaussian09量子化学程序包完成[14].
3 结果与讨论
3.1 几何构型的优化
采用密度泛函理论(DFT)对配合物1-4基态的几何构型进行了优化,计算结果列于表1.
表1 计算得到的配合物1-4主配体的部分二面角
Table1Computedandselecteddihedralangles(°)ofthemainligandsofcomplexes1-4

Comp.dihedralangledeviatedfrom180°1N(9)-C(11)-C(15)-C(21)C(11)-C(13)-C(14)-C(17)N(12)-C(13)-C(14)-C(16)C(13)-C(11)-C(15)-C(22)-138.6-152.5-146.6-144.741.427.533.435.32N(10)-C(12)-C(14)-C(18)N(11)-C(13)-C(20)-C(23)C(20)-C(15)-C(16)-C(21)C(14)-C(15)-C(16)-C(17)-0.2-0.1-0.100.20.10.103N(10)-C(12)-C(15)-C(18)N(11)-C(13)-C(14)-C(22)C(14)-C(16)-C(17)-C(19)C(15)-C(17)-C(16)-C(23)0.70.5179.4179.50.70.50.60.54N(10)-C(11)-C(14)-N(23)N(12)-C(13)-C(17)-N(18)N(18)-C(17)-C(16)-N(19)N(23)-C(14)-(15)-N(22)0.100-0.10.1000.1
由表1可以看出配合物1主配体的部分二面角偏离180°较多,其中最大的是N(9)-C(11)-C(15)-C(21)二面角偏离约41.4°,由此可知配合物1的平面性较差.而配合物2, 3和4的主配体的二面角基本上接近180°,其平面性较好.当配合物与DNA相互作用时,插入配体即主配体的平面性及共轭平面的大小是影响其与DNA相互作用的两个关键因素[15-17],配合物1的主配体平面性较差而且共轭平面面积较小,因此可以推测配合物1与DNA相互作用能力弱于其他三个配合物.
3.2 电子吸收光谱
在Ru(Ⅱ)多吡啶配合物的紫外可见吸收光谱中,在400-500nm范围内出现的强而宽的谱带,一般被认为金属到配体的电荷转移跃迁(MLCT).其中在200-300nm范围内最尖锐最强的谱带主要归因于辅助配体之间的π-π*跃迁,包含有少量的主配体之间的π-π*跃迁.表2列出了计算得到的配合物1-4在200-300nm范围内吸收谱带、相应的实验值,以及相关轨道跃迁,其中振子强度f>0.160,轨道归一化的贡献大于12%.表3列出了计算得到的配合物1-4在400-500nm范围内吸收谱带、相应的实验值,以及相关轨道跃迁,其中f>0.114,轨道归一化的贡献大于7%.
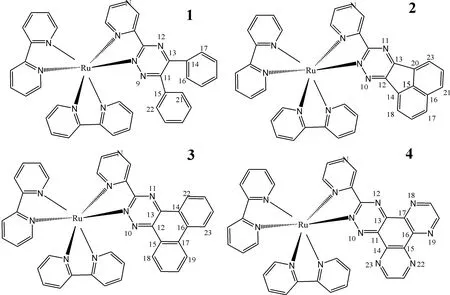
图1 配合物1-4的结构示意图及部分原子标签Fig. 1 Structural schematic diagrams of complexes 1-4 and the labels of partial atoms
表2 200-300nm范围内计算得到的配合物1-4的振子强度(f)、主要轨道跃迁贡献、模拟的吸收带及相应的实验值[18]
Table 2 Calculated oscillator strengths, main orbital transition contributions and simulated absorption bands of complexes 1-4 in the range of 200-300 nm as well as the experimental values[18]

Wavelength(nm)No.ExptSimu.Calc.faMajorcontributionCharacter1285.7277.03277.030.530HOMO-5→LUMO+3(38%)bHOMO-7→LUMO+2(24%)π(bpy)→π(bpy)*π(bpy)→π(bpy)*279.840.160HOMO-7→LUMO+3(30%)HOMO-6→LUMO+2(40%)π(bpy)→π(bpy)*π(bpy)→π(bpy)*2285.7280.29291.390.576HOMO-8→LUMO+1(85%)π(dta)→π(dta)*276.030.304HOMO-5→LUMO+4(46%)HOMO-4→LUMO+3(13%)HOMO-5→LUMO+2(15%)π(bpy)→π(dta)*π(bpy)→π(bpy)*π(bpy)→π(bpy)*278.020.258HOMO-4→LUMO+3(23%)HOMO-7→LUMO+2(12%)HOMO-6→LUMO+2(17%)HOMO-5→LUMO+4(16%)π(bpy)→π(bpy)*π(dta)→π(bpy)*π(bpy)→π(bpy)*π(bpy)→π(dta)*3282.1276.98276.980.721HOMO-5→LUMO+3(42%)HOMO-6→LUMO+2(33%)π(bpy)→π(bpy)*π(bpy)→π(bpy)*4275.04275.040.398HOMO-4→LUMO+4(14%)HOMO-6→LUMO+3(13%)π(bpy)→π(bpy)*π(dph)→π(bpy)*277.740.283HOMO-6→LUMO+4(26%)HOMO-1→LUMO+9(18%)HOMO-6→LUMO+3(17%)π(dph)→π(bpy)*π(bpy)→π(bpy)*π(dph)→π(bpy)*273.380.258HOMO-1→LUMO+10(19%)HOMO-4→LUMO+4(14%)π(bpy)→π(bpy)*π(bpy)→π(bpy)*
aOscillator strength;bThe percentage contributions to wave functions of excited states are given in parentheses
由表2可知,在200-300 nm光谱范围内,配合物1有两个f>0.160的强的跃迁.其中最强的谱带位于277.03 nm,其f=0.530,主要涉及到的跃迁是HOMO -5→LUMO+3 (38%)和 HOMO-7→LUMO+2 (24%)且都是π(bpy)→π(bpy)*.另一个在279.84 nm的谱带(f=0.160)主要涉及的跃迁是HOMO-6→LUMO+2 (40%)和HOMO-7→LUMO+3 (30%)也分别都是π(bpy)→π(bpy)*.因此,配合物1在285.7 nm的实验光谱可归因于理论上的277.03 nm和279.84 nm带的叠加,且计算拟合的吸收带波长为277.03 nm与实验值[18]285.7 nm吻合较好.对于配合物2,有三个f>0.160较强的谱带.在291.39 nm处有一个最强的吸收(f=0.576),对应于HOMO -8→LUMO+1 (85%)的跃迁,其特征是π(dta)→π(dta)*.在276.03 nm处是另一个较强的吸收谱带(f=0.304),对应的轨道跃迁及特征分别是HOMO-5 →LUMO+4 (46%)、π(bpy)→π(dta)*,HOMO-4→LUMO+3 (13%)、π(bpy)→π(bpy)*和HOMO-5→LUMO+2 (15%)、π(bpy)→π(bpy)*.在278.02 nm处是一个相对前两者较弱的吸收带,其振子强度为f=0.258,对应于HOMO-4→LUMO+3 (23%),HOMO-7→ LUMO +2 (12%),HOMO-6→LUMO+2 (17%)和HOMO-5→LUMO+4 (16%)的跃迁,相应的跃迁特征分别是π(bpy)→π(bpy)*、π(dta)→π(bpy)*、π(bpy)→π(bpy)*和π(bpy)→π (dta)*.因此配合物2在285.7 nm的实验光谱可归因于以上三个谱带(291.39 nm,276.03 nm和278.02 nm)的叠加,拟合的吸收带为280.29 nm与实验值285.7 nm相差5.41 nm.对于配合物3,最强的谱带位于276.98 nm(f=0.721),对应的轨道跃迁是HOMO-5→LUMO+3 (42%)和HOMO-6→LUMO+2 (33%),且都是π(bpy)→π(bpy)*,计算拟合的吸收带位于276.98 nm与实验值282.1 nm相差5.12 nm.对于配合物4,最强的谱带位于275.04 nm处(f=0.398),对应于HOMO-4→LUMO+4(14%)和HOMO-6 → LUMO +3(13%)的跃迁,其跃迁特征分别是π(bpy) →π(bpy)*和配合物4的主配体到辅助配体的跃迁π(dph) →π(bpy)*.另一个较强的谱带位于277.74 nm处(f=0.283),对应的轨道跃迁及特征分别是HOMO-6→LUMO+4(26%)、π(dph) →π(bpy)*,HOMO-1→LUMO+9(18%)、π(bpy)→ π(bpy)*和HOMO-6→LUMO + 3 (17%)、π(dph)→π(bpy)*.在273.38 nm处是一个相对前两者较弱的谱带(f=0.258),对应于 HOMO-1→LUMO+10(19%),HOMO-4 →LUMO+4 (14%)的跃迁,相应的跃迁特征都是π(bpy) →π(bpy)*.配合物4计算拟合的吸收带为275.04 nm.
表3 400 nm-500 nm范围内计算得到的配合物1-4的振子强度、主要轨道跃迁贡献、模拟的吸收带及相应的实验值
Table 3 Calculated oscillator strengths, main orbital transition contributions and simulated absorption bands of complexes 1-4 in the range of 400 nm - 500 nm as well as the experimental values
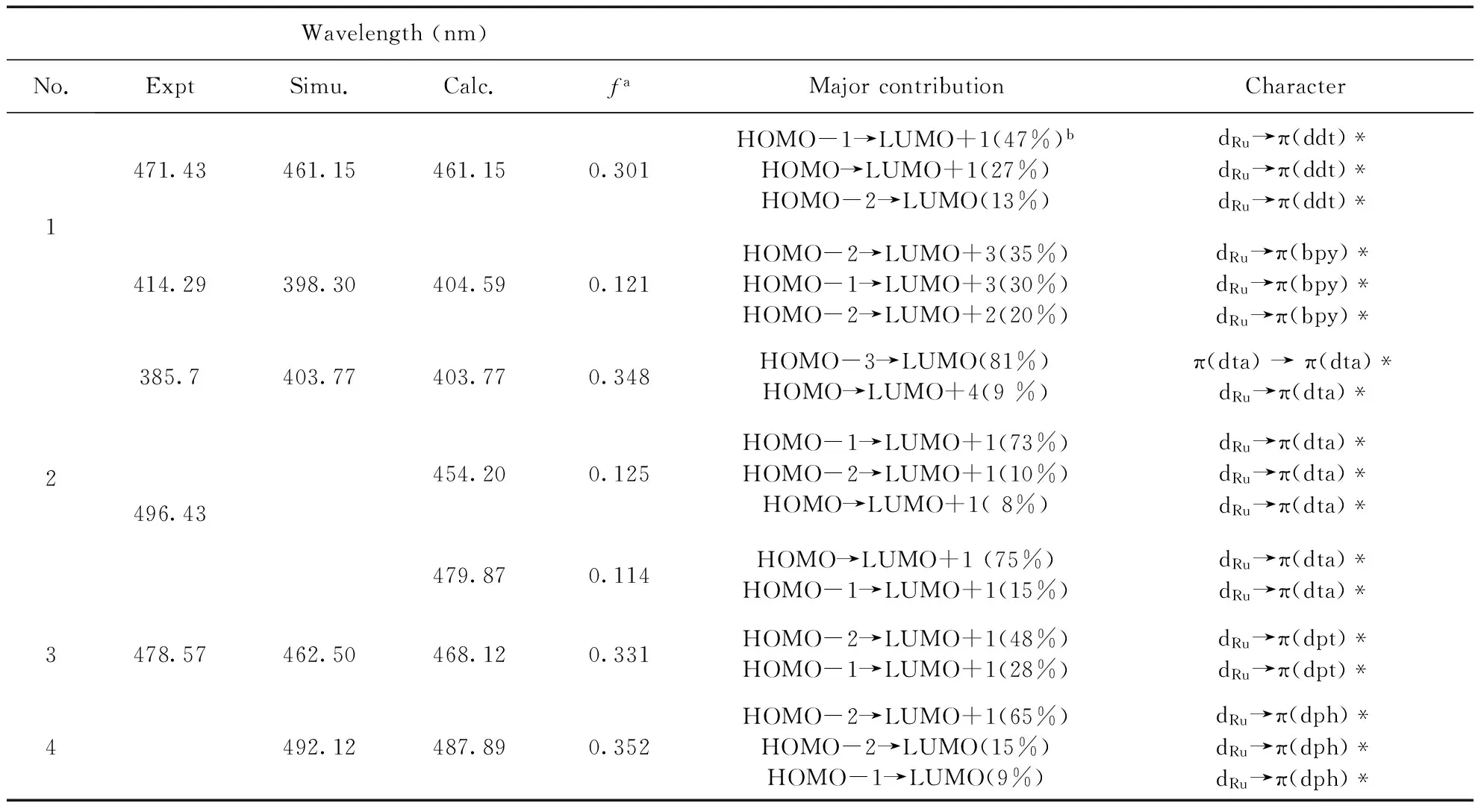
Wavelength(nm)No.ExptSimu.Calc.faMajorcontributionCharacter1471.43461.15461.150.301HOMO-1→LUMO+1(47%)bHOMO→LUMO+1(27%)HOMO-2→LUMO(13%)dRu→π(ddt)*dRu→π(ddt)*dRu→π(ddt)*414.29398.30404.590.121HOMO-2→LUMO+3(35%)HOMO-1→LUMO+3(30%)HOMO-2→LUMO+2(20%)dRu→π(bpy)*dRu→π(bpy)*dRu→π(bpy)*2385.7403.77403.770.348HOMO-3→LUMO(81%)HOMO→LUMO+4(9%)π(dta)→π(dta)*dRu→π(dta)*496.43454.200.125HOMO-1→LUMO+1(73%)HOMO-2→LUMO+1(10%)HOMO→LUMO+1(8%)dRu→π(dta)*dRu→π(dta)*dRu→π(dta)*479.870.114HOMO→LUMO+1(75%)HOMO-1→LUMO+1(15%)dRu→π(dta)*dRu→π(dta)*3478.57462.50468.120.331HOMO-2→LUMO+1(48%)HOMO-1→LUMO+1(28%)dRu→π(dpt)*dRu→π(dpt)*4492.12487.890.352HOMO-2→LUMO+1(65%)HOMO-2→LUMO(15%)HOMO-1→LUMO(9%)dRu→π(dph)*dRu→π(dph)*dRu→π(dph)*
aOscillator strength;bThe percentage contributions to wave functions of excited states are given in parentheses
由表3可知,在400-500 nm光谱范围内,配合物1有两个f>0.114的强的跃迁.其中最强的谱带位于461.15 nm,其f=0.301,主要涉及到的跃迁是HOMO-1→ LUMO+1(47%)、HOMO→LUMO+1(27%)和HOMO-2→LUMO(13%)且都是金属到主配体的跃迁dRu→π(ddt)*.另一个在404.59 nm的谱带(f=0.121)主要涉及的跃迁是HOMO-2→LUMO+3(35%)、HOMO-1→LUMO+3(30%)和HOMO-2→ LUMO +2(20%)且分别都是金属到辅助配体的跃迁dRu→π(bpy)*.第一个谱带拟合的吸收峰位于461.15 nm处与实验值471.43 nm相差10.28 nm,第二个位于398.30 nm处与实验值414.29 nm相差15.99 nm.对于配合物2,有三个f>0.114的强的跃迁.其中最强的谱带位于403.77 nm,其f=0.348,主要涉及到的跃迁是HOMO-3→ LUMO(81%)和HOMO→LUMO+4(9%),相应的跃迁特征分别是π(dta)→π(dta)*和dRu→π(dta)*.计算拟合的吸收峰位于403.77 nm处与实验值385.7 nm相差18.07 nm.另外,在454.20 nm(f=0.125)和479.87 nm(f=0.114)处有较强的吸收,对应的跃迁分别是HOMO-1→LUMO+1(73%)、HOMO-2→LUMO+1(10%)、HOMO→LUMO+1 (8%)和HOMO→LUMO+1(75%)、HOMO-1→LUMO+1(15%),且都是金属到主配体的跃迁dRu→π(dta)*.由于软件的局限性,实验上测得的在496.43 nm处的吸收峰未能拟合出,但是从图可以看出在496.43 nm左右应该有一个弱且宽的吸收锋.对于配合物3仅有一个最强的谱带,位于468.12 nm(f=0.331),对应的跃迁是HOMO-2→ LUMO+1(48%)和HOMO-1→ LUMO+1(28%),且都是dRu→π(dpt)*,计算拟合的吸收峰位于462.50 nm与实验值478.57 nm相差16.07 nm.对于配合物4,有一个比较强的谱带位于487.89 nm处(f=0.352),相应的跃迁分别是HOMO-2→LUMO+1(65%)、 HOMO-2 →LUMO (15%)和HOMO-1→LUMO(9%),跃迁特征都是金属到主配体的跃迁dRu-π(dph)*,计算拟合的吸收峰位于492.12 nm.
由以上分析可知,实验光谱图中峰的个数、峰的形状以及峰的位置与我们计算的吸收光谱差别都较小,因此可以说计算得到的电子吸收光谱与实验光谱吻合较好.从模拟的结果可以看出,200-300 nm范围内最尖锐最强的谱带主要归因于辅助配体之间的π-π*跃迁,在400-500 nm范围内出现的强而宽的谱带主要归因于d→π*跃迁,具有金属到配体的电荷转移性质(MLCT),这些谱带对研究钌配合物在DNA的发光性质中起着重要作用.从模拟的结果还可以看出,增加主配体的共轭面积使最大吸收波长蓝移(配合物3和配合物2相比),在主配体上增加电负性大的氮原子使最大吸收波长红移(配合物4和配合物3相比).
3.3 光裂解性质
计算得到的氧化还原电势值列于表4.由于配合物1-4激发态的氧化还原电势没有实验结果,为了比较计算结果和实验结果的吻合程度,对有实验结果的类似配合物[Ru(phen)3]2+基态和激发态的氧化还原电势进行了计算[19],计算结果也列于表4.由表4可知,基态[Ru(phen)3]2+的氧化还原电势分别为1.526 V、-1.065 V,分别接近实验值[20,21]1.27 V、-1.35 V;激发态的还原电位为0.914 V,接近实验值[20,21]0.70 V.计算得到的Ru(phen)3]2+的氧化还原势与实验值有一定的误差,这个误差可能是由于我们在计算中采用了CPCM模型(以水为溶剂)计算溶剂化能没有考虑氢键所致.该系列配合物的配体上含有电负性较大的氮原子能够与水分子形成强的氢键,目前我们所用的计算方法尚未考虑这种相互作用,因此这种方法计算的氧化还原电势还存在一定的误差.


图2 计算得到的配合物1-4的电子吸收光谱图(左边)及相应的实验光谱图(右边)Fig.2 Calculated (left) and experimental (right) absorption spectra of complexes 1-4
由表4还可以看出,配合物1-4的激发态还原电势(E*red)分别为1.143 V、0.697 V、1.098 V和1.239 V.表明E*red(4)>E*red(1)>E*red(3)>E*red(2), 钌配合物具有较高的激发态还原电势往往具有较强的DNA光裂解能力[22,23],因此可以预测配合物1-4光裂解DNA的能力(φ):φ(4)>φ(1)>φ(3)>φ(2),和实验结果[18]φ(3)>φ(2)>φ(1)不一致.从上面配合物1-3的几何结构可以看出,配合物2和3的主配体的平面性接近180°,而配合物1的主配体的平面性较差,当和DNA键合时,配合物1的主配体很难插进DNA碱基对,因此配合物1和DNA结合能力最差,这一点也得到实验证实[18].尽管配合物1的E*red很高,但其很难和DNA结合,其较高的E*red很难氧化DNA,因此可以预测,配合物1的光裂解DNA能力应该是最差的,配合物1-3光裂解DNA的能力:φ(3)>φ(2)>φ(1),得到合理的解释,设计的配合物4一方面具有较高的E*red,同时其主配体的平面性较好,从理论上预测配合物4具有较强的DNA光裂解能力.

表4 计算得到的配合物的氧化还原电势(V)
aOxidation potential of the complex in the ground state;bReduction potential of the complex in the ground state;cOxidation potential of the complex in the excited state;dReduction potential of the complex in the excited state
4 结 论
采用密度泛函理论优化了配合物1-4的几何结构,获得了配合物的电子结构性质,用TDDFT方法计算了四个钌多吡啶配合物在溶液中的电子吸收光谱, 计算的电子吸收光谱和实验结果吻合较好,并分析了配合物的主配体对电子吸收光谱性质的影响,对实验上测得的较强吸收带进行了详细解释.最后,计算了配合物1-4的氧化还原电势,并对配合物1-4的DNA光裂解能力进行了解释和预测.
[1] Barton J K. Metals and DNA: molecular left-handed complements [J].Science, 1986, 233 (4765): 727.
[2] Kelly S O, Homlin R E, Stemp E D,etal. Photoinduced electron transfer in ethidium-modified DNA duplexes: Dependence on distance and base stacking [J].J.Am.Chem.Soc., 1997, 119 (41): 9861.
[3] Kelly S O, Jackson N M, Hill M G,etal. Long-range electron transfer through DNA films [J].Angew.Chem.Int.Ed., 1999, 38 (7): 941.
[4] Farrell N. Biomedical uses and applications of inorganic chemistry. An overview [J].Coord.Chem.Rev., 2002, 232: 1.
[5] Gorling A. Density-functional theory for excited states [J].Phys.Rev. A, 1996, 54: 3912.
[6] Becke A D. A new mixing of Hartree-Fock and local density-functional theories [J].J.Chem.Phys., 1993, 98 (2): 1372.
[7] Hay P J, Wadt W R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg [J].J.Chem.Phys., 1985, 82 (1): 270.
[8] Wadt W R, Hay P J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi [J].J.Chem.Phys., 1985, 82 (1): 284.
[9] Mondal B, Chakraborty S, Munshi P,etal. Ruthenium-(Ⅱ)/-(Ⅲ) terpyridine complexes incorporating imine functionalities. Synthesis, structure, spectroscopic and electrochemical properties [J].J.Chem.Soc.Dalton.Trans., 2000, 2327.
[10] Das A K, Peng S M, Bhattacharya S. Ruthenium-mediated reduction of oximes to imines. Synthesis, characterization and redox properties of imine complexes of ruthenium [J].J.Chem.Soc.Dalton.Trans., 2000, 181.
[11] Reiss H, Heller A. The absolute potential of the standard hydrogen electrode: a new estimate [J].J.Phys.Chem., 1985, 89 (20): 4207.
[12] Barone V, Cossi M. Quantum calculation of molecular energies and gradients in solution by a conductor solvent model [J].J.Phys.Chem. A, 1998, 102 (11): 1995.
[13] Cossi M, Rega N, Scalmani G,etal. Energies, structures, and electronic properties of molecules in solution with the C-PCM salvation model [J].J.Comput.Chem., 2003, 24 (6): 669.
[14] Frisch M J, Trucks G W, Schlegel H B,etal. GAUSSIAN 09, Revision B.01, Gaussian, Inc. Wallingford, CT, 2010.
[15] Li J, Xu L C, Chen J C,etal. Density functional theory/time-dependent DFT studies on the structures, trend in DNA-binding affinities, and spectral properties of complexes [Ru(bpy)2(p-R-pip)]2+(R=-OH, -CH3, -H, -NO2) [J].J.Phys.Chem. A, 2006, 110 (26): 8174.
[16] Miao T F, Li S, Xu L C,etal. Theoretical studies on the DNA-intercalator properties of Co(Ⅲ) polypyridyl complexes [J].Comput.Theor.Chem., 2011, 976: 209.
[17] Li J, Chen J Can, Xu L C,etal. A DFT/TDDFT study on the structures, trend in DNA-binding and spectral properties of molecular“light switch”complexes [Ru(phen)2(L)]2+(L= dppz, taptp, phehat) [J].J.Org.Chem., 2007, 692: 831.
[18] Deng H, Cai J W, Xu H,etal. Ruthenium(Ⅱ) complexes containing asymmetric ligands: synthesis, characterization, crystal structure and DNA-binding [J].Dalton.trans., 2003, 325.
[19] Miao T F, Li S, Chen Q,etal. Probing DNA photocleavage efficiencies of Ru(Ⅱ) polypyridyl complexes: Theoretical calculation of redox potentials [J].Inorg.Chim.Acta, 2013, 407: 37.
[20] Herman L, Ghosh S, Defrancq E,etal. Ru(Ⅱ) complexes and light: molecular tools for biomolecules [J].J.Phys.Org.Chem., 2008, 21 (7-8): 670.
[21] Leveque J, Elias B, Moucheron C,etal. Dendritic tetranuclear Ru(Ⅱ) complexes based on the nonsymmetrical PHEHAT bridging ligand and their building blocks: synthesis, characterization, and electrochemical and photophysical properties [J].Inorg.Chem., 2005, 44 (2): 393.
[22] Miao T F, Li S, Chen J C,etal. Theoretical studies on DNA-photocleavage efficiencies and mechanisms of Ru(Ⅱ) polypyridyl complexes [J].J.Biol.Inorg.Chem., 2012, 17 (8): 1177.
[23] Miao T F, Li S, Chen J C,etal. Theoretical studies on DNA-photocleavage efficiencies of Ru(Ⅱ) polypyridyl complexes [J].Dalton.Trans., 2013, 42 (7): 2463.
Theoretical studies on DNA-photocleavage and spectral properties of a series of Ru(Ⅱ) complexes
WANG Na-Li1, WANG Qing-Qing1, MIAO Ti-Fang1,2, SUN Guo-Dong1, SONG Zhong-Ao1, SHI Jie1
(1. School of Chemistry and Materials Science, Huaibei Normal University, Huaibei 235000, China;2. State Key Laboratory of Theoretical and Computational Chemistry, Changchun 130023, China)
Theoretical studies of the electronic structures, DNA-photocleavage and spectral properties of a series of Ru (II) polypyridyl complexes 1-3 have been carried out using the density functional theory. Firstly, the oxidation redox potentials of complexes 1-3 were computed and the trend in the photocleavage abilities of these complexes was reasonably explained by their excited-state redox potentials. Secondly, complex 4 with a higher excited-state reductive potential was designed based on the electronic-structure properties of complexes 1-3, and the complex 4 with stronger DNA photocleavage ability was predicted. Finally, the electronic absorption spectra of complexes 1-4 were computed and simulated in aqueous solution using the time dependent DFT (TDDFT) method, and the calculated absorption spectra of these complexes are in satisfying agreement with the experimental ones. Moreover, the intense experimental absorption bands of these complexes are theoretically explained in detail and the effect of the main ligands on the electronic absorption spectra was also investigated.
Ru (Ⅱ) complexes; DFT method; DNA photocleavage; Spectral properties
2014-04-28
理论化学计算国家重点实验室基金(k2013-04);淮北市科技人才培育基金(20130305);大学生创新创业训练计划项目(201310373042, 201310373062, 201410373029)
王娜丽(1988—),女,河南杞县人,硕士研究生,主要从事生物无机理论和计算化学研究.
苗体方.E-mail: miaotifang@163.com
103969/j.issn.1000-0364.2015.10.005
O641.12
A
1000-0364(2015)05-0741-08
猜你喜欢
安徽化工(2022年1期)2022-02-15
防爆电机(2020年4期)2020-12-14
中华养生保健(2020年3期)2020-11-16
河北理科教学研究(2020年1期)2020-07-24
化工学报(2020年4期)2020-05-28
今日农业(2019年11期)2019-08-13
中国资源综合利用(2017年1期)2018-01-22
山东工业技术(2016年15期)2016-12-01
中国粮油学报(2016年5期)2016-01-23
高中生学习·高二版(2014年5期)2014-07-03
