
Illumina MiSeq高通量测序分析核桃内生细菌多样性
2015-03-15 12:32陈泽斌王定康徐胜光张永福彭声静
江苏农业学报 2015年5期
陈泽斌, 李 冰, 王定康, 余 磊, 徐胜光, 任 禛, 靳 松, 张永福,彭声静
(1.昆明学院农学院,云南 昆明 650214;2.云南省都市特色农业工程技术研究中心,云南 昆明 650214;3.中国科学院昆明动物研究所,云南 昆明 650223;4.昆明学院生命科学与技术系,云南 昆明 650214)
关于植物内生细菌,现已从多种健康植物的根、茎、叶、果实、种子中分离出包括革兰氏阳性菌和革兰氏阴性菌共计129种(隶属于54个属)[1]。植物内生细菌的优势种主要分布于假单胞属(Pseudommonas)、芽孢杆菌属(Bacillus)、肠杆菌属(Enterobacter)、土壤杆菌属(Agrobacterium)4 个属[2],研究较系统的植物有棉花、小麦和花生[3]。迄今还未发现不存在内生细菌的植物种类或器官。
核桃,又称胡桃、羌桃,为胡桃科植物,与扁桃、腰果、榛子并称为世界著名的“四大干果”,被誉为“万岁子”、“长寿果”[4-7]。核桃中86%的脂肪是不饱和脂肪酸,富含铜、镁、钾、维生素B6、叶酸和维生素B1,也含有纤维、磷、烟酸、铁、维生素B2和泛酸,能破血祛瘀、润燥滑肠,并能防癌抗癌、补脑[8]。中国核桃的分布很广,主要产区在云南、陕西、山西、四川、河北、甘肃、新疆、安徽等省(区)。
已有研究结果表明植物内生细菌的种类组成随植物的品种、生长期、器官的不同而异[9]。目前关于核桃内生菌多样性的研究均采用传统培养方法,研究的部位集中于核桃植株的根、茎、叶[10-11],而对于可供食用的部分——核桃仁这一器官的内生细菌研究未见报道。本研究拟采用Illumina MiSeq第二代测序技术对核桃仁内生细菌种类组成进行分析,该方法相较于传统的纯培养及以16SrDNA为基础的非培养方法,能够产生覆盖深度更大的数据量,检测到纯培养和非培养技术未能发现的低丰度植物内生细菌种类,为丰富植物微生态学理论及基因工程菌的研究奠定基础。
1 材料与方法
1.1 样品来源
供试材料为云南省林业科学院经济林研究所提供的核桃品种(无性系)——细香核桃。
1.2 样品消毒
将新鲜核桃仁用自来水冲洗干净后用75%的乙醇浸泡1 min,无菌水冲洗3次,用无菌滤纸吸干表面水分。消毒效果检验参照文献[12]。
1.3 核桃仁总DNA提取
[13]的方法提取核桃仁总DNA,并用0.8%的琼脂糖凝胶电泳检查DNA的纯度和浓度。取适量的样品于离心管中,使用无菌水稀释样品至 1 ng/μl。
1.4 16SrDNA-V4区的PCR扩增
以稀释后的基因组DNA为模板,使用带Barcode的16SrDNA-V4区特异引物515F(5'-GTTTCGGTGCCAGCMGCCGCGGTAA-3')和806R(5'-GATCAGGGACTACHVGGGTWTCTAAT-3'),并用高效和高保真酶(New England Biolabs公司的Phusion®High-Fidelity PCR Master Mix with GC Buffer)进行PCR,确保扩增效率和准确性。
1.5 PCR产物的混样和纯化
PCR产物用2%的琼脂糖凝胶进行电泳检测,目的条带用Qiagen公司提供的胶回收试剂盒回收产物。
1.6 文库构建和测序
用TruSeq®DNA PCR-Free Sample Preparation Kit建库试剂盒进行文库构建,构建好的文库经过Qubit和 QPCR 定量,合格后使用 Miseq 测序[14-17]。测序委托北京诺禾致源生物信息科技有限公司完成。
1.7 生物信息学分析
测序得到的原始数据存在一定比例的干扰数据,为了使信息分析的结果更加准确、可靠,首先对原始数据用FLASH软件拼接、Qiime软件过滤、UCHIME Algorithm软件去除嵌合体后,得到有效数据(Effective tags)[18],剔除宿主叶绿体和线粒体序列,然后用Uparse软件对有效数据在97%水平上进行操作分类单元(Operational taxonomic unit,OTU)聚类,并利用Greengene数据库进行物种注释[19],利用Mothur软件做稀释曲线分析,通过对OTUs进行丰度和α-多样性分析,得到微生物群落结构组成[20-22]。
2 结果与分析
2.1 序列长度分布
在核桃仁样品总DNA的16S rDNA V4区中测得的63 190条序列,双末端读长(PE,Paired-end reads)分布在 189~359 bp范围内,长度为249 bp的序列最多,有62 621条(表1)。从序列长度的分布来看,与16SrDNA-V4区序列长度大致吻合。
2.2 OTUs数目统计及物种注释分析
核桃仁样品总DNA的16S rDNA V4区中共测得63 183条标记(Tags,过滤后得到的拼接序列数),分为103个OTUs(97%的序列相似性),包括61 980条获得分类信息的标记,1 203条低频标记(图1),被注释到11个属(表2)。从OTUs丰度聚类图(图2)来看,相对丰度为土壤杆菌属(Agrobacterium)>鞘氨醇单胞菌属(Sphingomonas)=盐单胞菌属(Halomonas)。

表1 核桃仁内生细菌16Sr DNA V4区序列长度的数量分布Table 1 Distribution of sequence length of 16Sr DNA V4 region of endophytic bacteria in walnut kernel

图1 样品中内生细菌序列的标记和OTUs的数目Fig.1 The quantities of tags and OTUs of endophytic bacteria in the sample
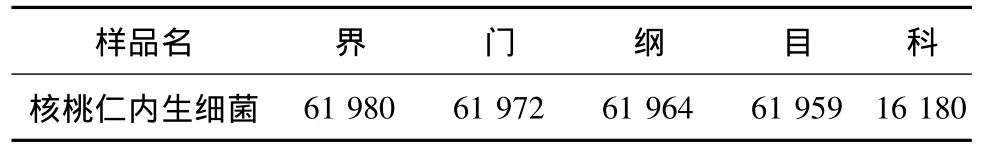
表2 核桃仁内生细菌16S r DNA序列各分类水平上的序列数目Table2 Sequence number at each classification level of 16S r DNA of endophytic bacteria in walnut kernel
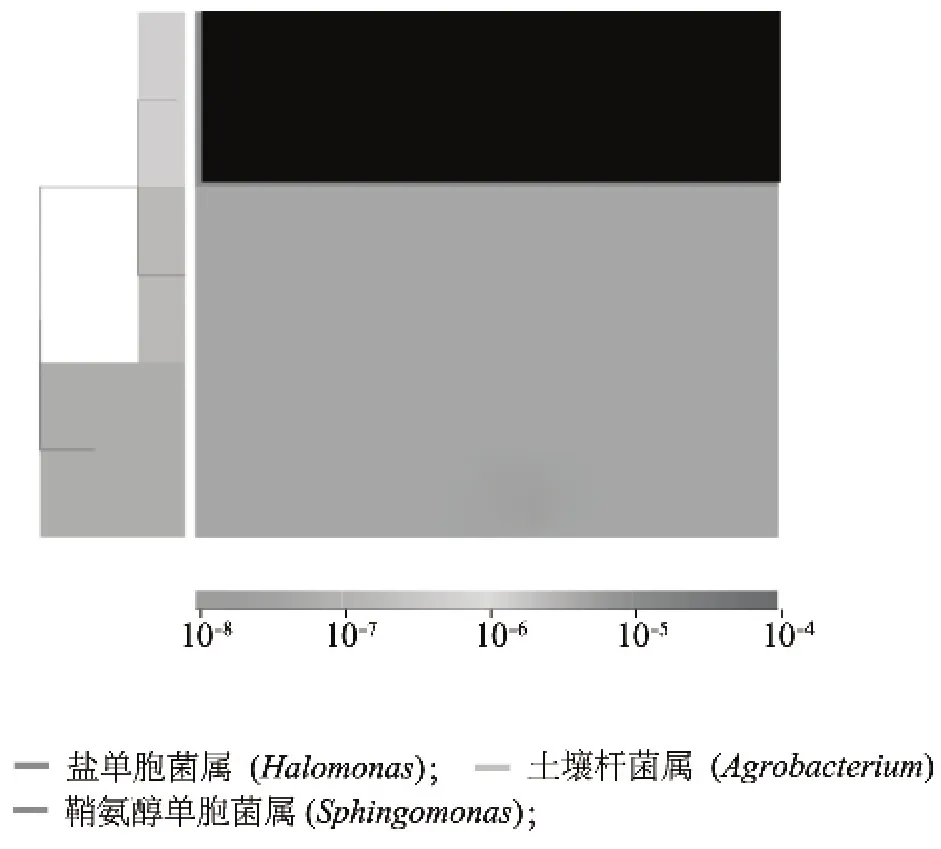
图2 核桃仁内生细菌16S r DNA序列OTUs丰度聚类图Fig.2 Abundance clustering of OTUs of 16S r DNA of endophytic bacteria in walnut kernel
2.3 样品中内生细菌物种分类树
从门的分类水平看,核桃仁内生细菌主要分布在变形菌门(Proteobacteria);从纲的分类水平看,核桃内生细菌在 γ-变形菌纲(Gammaproteobacteria,27.27%)、 α-变 形 菌 纲 (Alphaproteobacteria,72.73%)中均有分布;从目的分类水平看,核桃内生细菌在鞘脂杆菌目(Sphingobacteriales,27.27%)、根瘤菌目(Rhizobiales,45.45%)、海洋螺菌目(Oceanospirillales,27.27%)中均有分布;从科的分类水平看,核桃内生细菌在根瘤菌科(Rhizobiaceae,45.45%)、鞘 脂 单 胞 菌 科 (Sphingomonadaceae,27.27%)、盐单胞菌科(Halomonadaceae,33.33%)中均有分布;从属的分类水平看,核桃内生细菌在鞘氨醇单胞菌属(Sphingomonas,27.27%)、盐单胞菌属(Halomonas,27.27%)、土壤杆菌属(Agrobacterium,45.45%)中均有分布。由此可见,鞘氨醇单胞菌属、盐单胞菌属、土壤杆菌属是核桃内生细菌的优势种群(图3)。
2.4 α-多样性分析
随着测序数量的增加,稀释曲线(图4)、Chao1指数曲线(图5)、Shannon指数曲线(图6)的斜率在0~10 000的测序数量范围内逐渐上升,后趋向平坦并进入平台期,说明再增加测序数量也不会产生新的OTU,也表明本研究测序数据量足以反映样品中绝大多数的微生物信息。

图3 核桃仁中内生细菌的分类Fig.3 Classification of endophytic bacterial species in sample walnut kernel

图4 稀释曲线Fig.4 Rarefaction curve

图5 Chao1指数曲线Fig.5 Chao1 index curve

图6 Shannon指数曲线Fig.6 Shannon index curve
3 讨论
16SrDNA很早就应用在微生物菌种鉴定上,但利用高通量测序进行16S rDNA分析最早出现在2006年,Sogin等[22]应用该技术对深海微生物群落进行了分析,随后该技术在肠道微生物领域得到了广泛应用[23]。目前该技术应用于植物内生微生物的研究尚未见报道。本研究首次将近几年迅速发展并成为主流的Illumina MiSeq第二代测序技术应用于植物内生细菌研究,克服了传统分子生物学方法通量低的缺陷,从基因组水平上解析微生物群落结构,突破了很多厌氧内生微生物尚不能被分离培养的技术瓶颈[24-25],检测到以往没有检测到的同样扮演着重要角色的低丰度植物内生细菌种类,丰富了植物内生细菌资源,更准确地反映了植物内生微生物中不同丰度菌群的组成和比例。
就植物组织而言,叶绿体和线粒体与细菌在系统发育上具有高度的同源性[26]。在对核桃内生细菌的16SrDNA-V4变异区序列进行Illumina MiSeq高通量测序后,发现部分序列归属于宿主叶绿体和线粒体,存在一定比例的宿主污染现象,为了使细菌多样性的分析结果更加准确、可靠,本研究剔除了宿主叶绿体和线粒体序列。但本研究只选择了16S rDNA-V4区作为测序区域,未来研究应在不同高通量测序平台上评估样品的复杂程度以及宿主的污染情况,选择合理的测序区域或设计特异性更高的引物,制定更好的测序策略,以避免线粒体和叶绿体的干扰。
第一代Sanger测序由于其读长长、准确率高,能根据测得的全长16S rDNA序列注释到种,但第一代Sanger测序通量较低,大规模测序成本高。Illumina MiSeq高通量测序技术,一般每个样品可以保证测定40 000~60 000个序列数,覆盖深度非常大,对物种多样性的分析十分有利,但是高通量测序读长短,不可能将16S rDNA的9个可变区全部测序,所以往往只选择1~3个可变区作为测序区域,单端测序长度为250 bp,双端测序长度为500 bp,测序片段越短,后续对序列进行物种注释时的分辨度越低,只能将内生细菌注释到属的分类水平。
本研究发现核桃内生细菌主要分布于鞘氨醇单胞菌属、盐单胞菌属、土壤杆菌属,这3个属是核桃内生细菌优势菌属。已有研究结果表明,鞘氨醇单胞菌属细菌的底物广泛,从多环芳烃类化合物、聚乙烯醇等高聚物到简单无机物氮都能利用,某些种属还能产生有价值的生物高分子(如β-胡萝卜素、结冷胶)[27]。说明核桃内生细菌可为复杂有机物的降解提供良好的微生物来源。目前关于盐单胞菌属细菌功能、代谢的研究尚未见报道,其作为内生细菌的主要类群尚属首次报道,未来应进一步加强对其功能和代谢的研究。
参考文献:
[1] 赵 旭,常思静,景春娥,等.我国植物内生菌研究进展[J].中国沙漠,2010,30(1):87-91.
[2] 徐亚军.植物内生菌资源多样性研究进展[J].广东农业科学,2011(24):149-152.
[3] 胡桂萍,郑雪芳,尤民生,等.植物内生菌的研究进展[J].福建农业学报,2010,25(2):226-234.
[4] 王 伟,翟梅枝,徐文涛,等.核桃内生菌研究Ⅰ核桃内生菌的分离及代谢产物活性研究[J].西北农业学报,2008,17(1):77-81.
[5] 高美娟,任建军,师光禄,等.核桃青皮复配营养液对设施草莓的杀螨及营养效果[J].江苏农业学报,2013,29(6):1320-1325.
[6] 刘广勤,俞卫东,曹仁勇,等.薄壳山核桃食药用价值及加工利用研究进展[J].江苏农业科学,2014,42(12):306-303.
[7] 李 丽,翟梅枝,杨 惠,等.核桃叶部内生真菌发酵产物抑菌活性及 GC-MS 分析[J].江苏农业科学,2014,42(10):292-295.
[8] 李晓红,傅本重,麻春花,等.不同表面消毒方法对核桃叶内生菌分离效果的比较[J].中国农学通报,2012,28(28):163-168.
[9] 高智辉,翟梅枝,王云果,等.核桃内生真菌的分离鉴定[J].西北林学院学报,2012,27(5):121-123.
[10]李 丽,翟梅枝,杨 惠,等.核桃内生真菌G3的分子鉴定及其生物学特性研究[J].中国农学通报,2013,29(10):35-39.
[11]王建文,陈华红,段亚霞,等.楚雄核桃内生真菌分离及抗菌活性研究[J].楚雄师范学院学报,2013,28(3):70-75.
[12]陈泽斌,代方平,寸林江,等.烟草内生细菌分离方法的优化研究[J].中国烟草学报,2014,20(1):90-95,102.
[13]陈泽斌,夏振远,雷丽萍,等.非培养方法解析烟草根部内生细菌的群落结构[J].华北农学报,2012,27(1):201-209.
[14] CAPORASO JG,LAUBER CL,WALTERSW A,et al.Global patterns of 16SrRNA diversity at a depth of millions of sequences per sample[J].Proceedings of the National Academy of Sciences,2011,108(Suppl 1):4516-4522.
[15] YOUSSEF N,SHEIK C S,KRUMHOLZ L R,et al.Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys[J].Applied and Environmental Microbiology,2009,75(16):5227-5236.
[16] HESSM,SCZYRBA A,EGAN R,et al.Metagenomic discovery of biomass-degrading genes and genomes from cow rumen[J].Science,2011,331(6016):463-467.
[17] LUO C,TSEMENTZI D,KYRPIDES N,et al.Direct comparisons of Illumina vs.Roche 454 sequencing technologies on the same microbial community DNA sample[J].PloS One,2012,7(2):e30087.
[18] EDGAR R C,HAASB J,CLEMENTE JC,et al.UCHIME improves sensitivity and speed of chimera detection[J].Bioinformatics,2011,27(16):2194-2200.
[19] EDGAR R C.UPARSE:highly accurate OTU sequences from microbial amplicon reads[J].Nature Methods,2013,10(10):996-998.
[20] WANG QL,GARRITY GM,TIEDJE JM,et al.Naive Bayesian classifier for rapid assignment of rRNA sequences into thenew bacterial taxonomy[J].Applied and Environmental Microbiology,2007,73(16):5261-5267.
[21] DESANTIST Z,HUGENHOLTZ P,LARSEN N,et al.Greengenes,a chimera-checked 16S rRNA gene database and workbench compatible with ARB[J].Applied and Environmental Microbiology,2006,72(7):5069-5072.
[22] SOGIN M L,MORRISON H G,HUBER JA,et al.Microbial diversity in the deep sea and the underexplored‘rare biosphere’[J].Proceedings of the National Academy of Sciences,2006,103(32):12115-12120.
[23]南春燕,马雅军,徐建农,等.中华按蚊幼虫肠道细菌宏基因组的组成研究[J].中国寄生虫学与寄生虫病杂志,2013,31(2):114-119.
[24] THOLOZAN JL,CAPPELIER J M,TISSIER JP,et al.Physiological characterization of viable-but-nonculturable campylobacter jejuni cells[J].Appl Environ Microbiol,1999,65(3):1110-1116.
[25] AMMANN R R,LUDWIG W,SCHLEIFER K H.Phylogenetic identification and in situ detection of individual microbial cells without cultivation[J].Microbiol Rev,1995,59(1):143-169.
[26]胡 楷,吴庆书.单细胞生物进化研究的进步[J].遗传,2002,24(1):104-110.
[27]胡 杰,何晓红,李大平,等.鞘氨醇单胞菌研究进展[J].应用与环境生物学报,2007,13(3):431-437.
猜你喜欢
小雪花·成长指南(2022年1期)2022-04-09
基层中医药(2020年2期)2020-07-27
湖北农机化(2020年4期)2020-07-24
世界农药(2019年4期)2019-12-30
饮食科学(2019年5期)2019-11-21
今日农业(2019年11期)2019-08-15
乡村地理(2018年2期)2018-09-19
中成药(2017年9期)2017-12-19
中华老年口腔医学杂志(2016年2期)2017-01-15
现代检验医学杂志(2015年1期)2015-02-06
