
遗传性球形红细胞增多症合并肝内胆汁淤积致严重高胆红血症1例*
2015-03-15 02:33陈源文曹海霞范建高
实用肝脏病杂志 2015年3期
周 达,陈源文,曹海霞,范建高
遗传性球形细胞增多症(Hereditary spherocytosis,HS)是一种常染色体显性遗传性疾病,临床上主要表现为溶血性贫血伴黄疸,以间接胆红素升高为主,很少合并肝内胆汁淤积和极度的高胆红素血症。我们报道1例罕见的遗传性球形细胞增多症伴有直接胆红素极度升高和肝内胆汁淤积患者,对疾病进程中一些特殊的临床表现发生的原因进行了分析。
1 病例摘要
患者女性,49岁,会计。因“反复腹痛伴尿黄皮肤黄40天”于2013年10月10日入院。缘于2013年8月29日无明显诱因下出现中上腹和两侧季肋部持续性胀痛,恶心,呕吐数次,为胃内容物,伴乏力,同时发现全身皮肤和眼黄,尿色加深,呈酱油色,大便如常,无发热。患者2月前体检发现有胆囊结石。患者出生时黄疸较深,幼年即被诊断为“溶血性贫血”,平时血红蛋白在80 g/L左右。查体:全身皮肤、巩膜黄染,可见肝掌,无蜘蛛痣。K-F环阴性,心肺听诊无异常发现。腹平软,无腹壁静脉曲张。右上腹轻压痛,无反跳痛,未触及肿块。肝上界正常,Murphy征阴性,肝肋下未触及,脾肋下5 cm,质中,无触痛。移动性浊音阴性,肠鸣音正常。辅助检查:肝功能和血常规变化见表1和表2,肝炎病毒标记物均阴性,血铜蓝蛋白和尿铜正常,甲状腺激素正常,抗核抗体(ANA)、抗线粒体抗体(AMA)、抗中性粒细胞胞质抗体(ANCA)均阴性,血清IgG和IgM正常,IgA为8.7 g/L,IgG4为0.362 g/L(参考值 0.08~1.4 g/L),结合珠蛋白 0.07 g/L。直接抗人球蛋白试验阴性。红细胞脆性实验:球形红细胞7.3%,网织红细胞 8.1%。上腹部MRI提示胆囊结石,胆囊积液,肝多发囊肿,脾肿大(重度),未见肝内外胆管扩张(图1 A、B和C)。腹部B超提示胆囊多发结石(较大者直径为10 mm),胆囊颈部结石(直径为8 mm),胆囊炎,脾肿大(171×54 mm)。肝活检提示肝小叶结构存在,局部可见肝细胞气球样变,肝细胞再生明显,肝细胞内及毛细胆管淤胆,局部可见微泡性脂肪变和嗜酸性小体,肝窦内可见D-PBS阳性的蜡质样细胞,汇管区轻度纤维化,个别中性粒细胞和少量淋巴细胞浸润(图2 A和B)。免疫组化检测发现:CD34+、CK19+、CK7+,HBcAg和 HBsAg阴性,D-PAS阳性,MASSON染色正常,普鲁士蓝染色阳性,网状纤维染色正常。

表1 患者肝功能和血清激素水平的变化
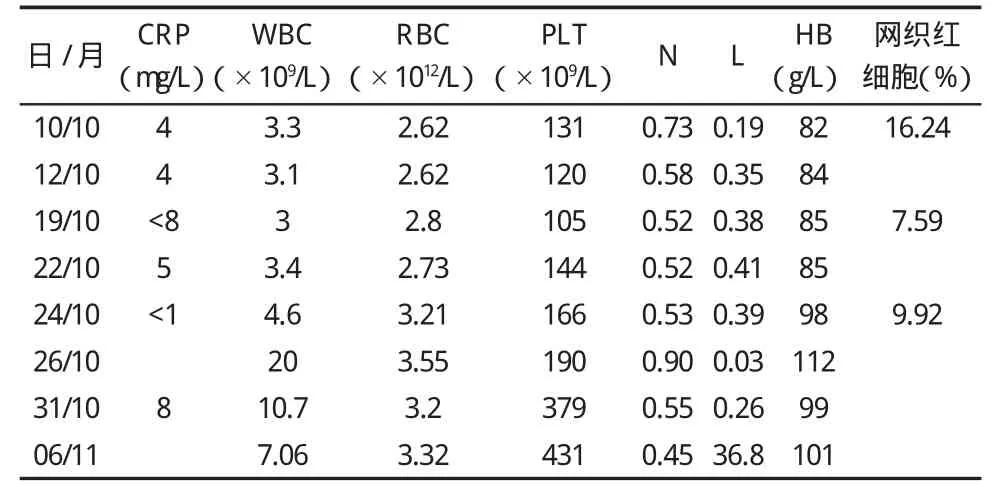
表2 血常规和C反应蛋白的变化

图1 腹部MRI表现 胆囊结石和胆囊积液明显(A和B),
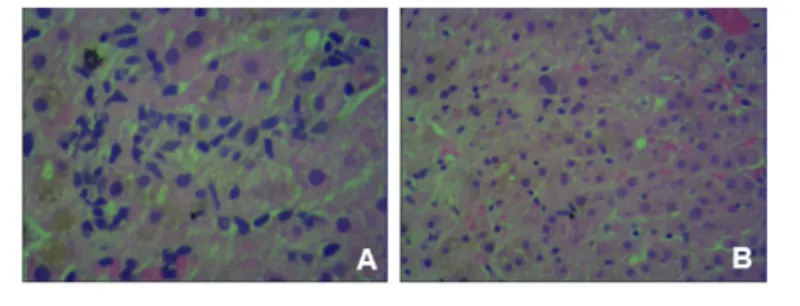
图2 肝组织学表现 提示毛细胆管淤胆(HE,200×)
患者入院时,考虑严重高胆红素血症和溶血性贫血原因不明,肝内胆汁淤积可能,予以丁二磺酸腺苷蛋氨酸(思美泰)保肝降黄及对症支持治疗;因患者腹痛,考虑胆道感染可能,予以左氧氟沙星联合甲硝唑抗感染对症治疗。经完善溶血性贫血相关检查后,发现患者球形红细胞增多(7.3%)符合遗传性球形细胞增多症诊断标准,但仍不能解释患者如此严重的高胆红素血症。肝功能检查提示直接胆红素/总胆红素>50%,且肝活检提示肝内胆汁淤积,以及腹部MRI和B超均未见肝外梗阻依据,考虑重度肝内胆汁淤积为严重高胆红血症的主要原因,故予以甲强龙(40 mg,bid)口服治疗 3 d,甲强龙(40 mg,qd)治疗 3 d,患者血清总胆红素由649.3μmol/L下降至178μmol/L,此后仍维持在高水平,不再断续下降。鉴于患者遗传性球形红细胞增多症及胆囊结石诊断明确,右上腹痛时常发作,除高胆红素血症外,患者精神状态、食欲均良好,白蛋白、凝血酶原时间等肝功能指标基本正常,经多学科会诊,于10月25日行胆囊及脾切除术,术中探查见肝脏无硬化,腹腔内无积液,脾脏肿大,约20 cm×16 cm×6 cm,胆囊大小7 cm×3 cm×3 cm,与周围组织无明显粘连,解剖见胆囊管直径0.3 cm,胆总管直径0.6 cm,胆囊壁厚约0.3 cm,内见直径0.4~0.6 cm小结石数枚。术后病理学诊断:淤血性脾肿大,慢性胆囊炎,胆囊肌腺症(胆囊粘膜肥厚性增生深入肌层),胆固醇性息肉形成,胆石症。术后患者肝功能、血常规快速恢复正常,于11月7日出院。出院后2月随访复查,肝功能正常。
2 讨论
遗传性球形细胞增多症是一种红细胞膜异常的溶血性贫血,主要为红细胞5种膜蛋白中一种或多种蛋白的缺陷导致,属常染色体显性遗传病,临床表现主要为贫血、黄疸和脾肿大,常见有胆结石等[1],其诊断主要依靠典型上述临床症状及相关实验室检查(以游离胆红素增高、网织红细胞增高,外周血涂片球形红细胞>7%~10%,Coombs实验阴性,红细胞渗透脆性增加),如合并明确家族史,或红细胞膜缺陷分析或基因检查发现膜蛋白缺陷更有利于诊断,其治疗方面除了内科对症治疗外,对溶血明显(重度贫血、总胆红素>34 μmol/L)患者需行脾切除治疗,效果显著[1,2]。
本例患者诊断HS无异议。然而,HS一般以间接胆红素升高为主,总胆红素一般<85μmol/L),而本例患者入院时直接胆红素升高显著,且占总胆红素的54.9%,总胆红素最高达649μmol/L,这在国内外鲜有报道,显然不能用单纯HS来解释,也不符合急性溶血危象的表现。在影像学检查排除明显的肝外梗阻性胆汁淤积后,结合患者AST、ALT异常,直接胆红素/总胆红素>50%,肝内胆汁淤积的诊断基本成立,而肝穿刺见肝细胞和毛细胆管淤胆则基本确定为肝内胆汁淤积[3]。因此,有可能为长期HS继发肝内淤胆,但是否仍存在其他肝内胆汁淤积合并症需要慎重考虑。例如,国外曾报道过一例类似患者,最后考虑胆道内结石通过不畅所致一过性淤胆,行内镜下逆行胰胆管造影术(ERCP)治疗后胆汁淤积显著改善[3]。本例患者术前存在腹痛,术后病理学检查提示胆囊内有小结石数枚,同时HS最常见并发症即为胆结石[2],故一过性胆石通过困难引发淤胆也不能排除,且可能系患者反复腹痛的原因。
肝细胞对胆盐的处理过程复杂,其中涉及多种酶类及离子泵。在缺氧、免疫炎症等情况下,这些酶或离子泵的功能可能受到抑制,加之有大量胆红素需要合成和排泄时,这种平衡可能被打破,从而导致严重的肝内胆汁淤积。此前国内有过类似报道1例[4],考虑淤胆为HS继发脾功能亢进,使过量破损红细胞入血激发肝组织发生免疫炎性反应所致。严重肠道感染、胆道及周围炎症诱发与肝功能代偿状态不一致的严重胆汁淤积在临床也常见。这些感染因素可能可以解释患者常在腹痛症状发作后黄疸迅速加深,但其它肝功能指标却始终保护在基本正常水平的现象。
除上述原因外,国外有过报道一例HS合并遗传异常导致淤胆,因ABCB11基因突变而致良性复发性肝内胆汁淤积2型(Benign recurrent intrahepatic cholestasis-2,BRIC-2)[5]。该类疾病主要为编码胆汁排泄的基因突变而致淤胆,主要与ATP8B1、ABCB11、ABCB4三个基因有关。其中BRIC1型、家族性进展性肝内胆汁淤积(Progressive familial intrahepatic cholestasis,PFIC)1型主要为 ATP8B1缺陷引起;BRIC2、PFIC2主要为ABCB11缺陷引起;PFIC3型主要为ABCB4缺陷引起。一般BRIC发病可在各年龄段,症状较轻,以阵发性发作淤胆、黄疸、瘙痒为主,对肝脏损害轻,可由各种因素诱发(如感染、发热、药物、妊娠等)。该病例从病史、临床表现及实验室检查分析更倾向于BRIC可能,但对该患者ATP8B1、ABCB11两个基因外显子(各28个外显子)进行突变位点检测,仅发现单核苷酸多态性SNP(ATP8B1的27号外显子第54个碱基G>A、第77个碱基C>T;ATP8B1的16号外显子第99个碱基A>G;ABCB11的13号外显子第23个碱基T>C;ABCB11的24号外显子第28个碱基A>G)。因为仅对外显子检测,也有可能是内含子异常所致,因此无阳性突变不能完全排除基因问题。
通过该病例分析,应该认识到,HS确诊患者可以合并严重肝内胆汁淤积,但较罕见,应尽量排除其他淤胆原因,及时制定针对性的诊疗方案;对于肝病与消化专业医师而言,在严重胆汁淤积与患者肝功能代偿的其它指标不一致时,不要轻易作出肝衰竭的诊断而应该努力寻找其它合并的因素,特别是溶血,以及胆道周围的感染,而后者常常会诱发或加重可能存在的溶血。对于怀疑肝内胆汁淤积时,肝穿刺病理活检虽为有创性检查,但有助于病理诊断,尤其在那些病因不明,且无肝活检禁忌证的患者。当然,基因检测对于部分患者的鉴别诊断可能是必要的。
[1]Bolton-Maggs PH,Langer JC,Iolascon A,et al.Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update.Br J Haematol,2012,156(1):37-49.
[2]Perrotta S,Gallagher PG,Mohandas N.Hereditary spherocytosis.Lancet,2008,372(9647):1411-1426.
[3]Kalinke L,Rashid M.A cholestatic diagnostic dilemma.BMJ Case Rep,2013,2013.
[4]赵彩彦,王玮,刘英辉,等.遗传性球形红细胞增多症合并重度肝内胆汁淤积和继发性血色病.中华肝脏病杂志,2010,18(7):552-553.
[5]Wree A,Canbay A,Muller-Beissenhirtz H,et al.Excessive bilirubin elevation in a patient with hereditary spherocytosis and intrahepatic cholestasis.Z Gastroenterol,2011,49(8):977-980.
猜你喜欢
中国现代医生(2022年19期)2022-11-04
电子科技大学学报(2022年5期)2022-10-29
保健医苑(2022年5期)2022-06-10
昆明医科大学学报(2022年4期)2022-05-23
基层中医药(2021年8期)2021-11-02
中国生殖健康(2020年4期)2021-01-18
肝博士(2020年5期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
华北水利水电大学学报(社会科学版)(2015年3期)2015-02-28
肝博士(2015年2期)2015-02-27
