
替米沙坦对血管紧张素Ⅱ所致血管内皮细胞游离钙离子的抑制作用
2015-03-11 01:14黄汉辉郑师明荆冬
中国医药导报 2015年3期
黄汉辉++++++郑师明++++++荆冬冬++++++刘少志++++++梁+++颖++++++关秉恩+严鹏科
[摘要] 目的 探讨血管紧张素Ⅱ(AngⅡ)对细胞钙库的应激效应及替米沙坦对此的拮抗作用。 方法 预防性实验分组:正常对照组、AngⅡ 0.1 μmol/L组和替米沙坦60、300、1000 μg/L预处理组;治疗性实验分组:正常对照组、AngⅡ 0.1 μmol/L组和AngⅡ+替米沙坦60、300、1000 μg/L组。分别检测各组试验前胞内钙离子浓度,记为0 h。体外培养的人大动脉血管内皮细胞预防性给予替米沙坦60、300、1000 μg/L培育30 min后,加入AngⅡ 0.1 μmol/L;或AngⅡ 0.1 μmol/L培育30 min后,再加入替米沙坦60、300、1000 μg/L,分别于共同作用0.5、2、8和24 h采用激光共聚焦扫描显微镜成像法检测胞浆游离钙离子浓度。 结果 预防性实验:与正常对照组相比,替米沙坦60、300、1000 μg/L作用细胞30 min对胞浆游离钙离子浓度无明显影响,加入AngⅡ 0.1 μmol/L后,胞浆游离钙离子浓度立即(0 h)显著升高,均显著高于正常对照组(P < 0.05),但显著低于AngⅡ组,并且替米沙坦预处理浓度越高抑制作用越强(P < 0.05),作用时间对此无影响。治疗性实验:AngⅡ 0.1 μmol/L先诱导30 min后,再加入替米沙坦60 μg/L对升高的胞浆游离钙离子浓度无抑制作用;替米沙坦300 μg/L作用2 h或替米沙坦1000 μg/L作用0.5 h才显示其抑制作用(P < 0.05),但均显著高于同一时间点的正常对照组。替米沙坦的作用时间对此无影响。 结论 AngⅡ能诱导内质网钙库向胞浆释放游离钙离子,而替米沙坦能有效拮抗AngⅡ对内质网的应激作用及促使胞浆游离钙离子重新被内质网重吸收。预防性给予替米沙坦的拮抗作用较治疗性给予的拮抗作用显著。
[关键词] 替米沙坦;血管内皮细胞;血管紧张素Ⅱ;钙离子
[中图分类号] R972.4 [文献标识码] A [文章编号] 1673-7210(2015)01(c)-0011-05
高血压、动脉硬化、冠心病等心血管系统疾病的发生发展过程中均存在着以内皮细胞所分泌的一氧化氮(NO)减少而致血管舒张反应下降为主要特征的血管内皮系统功能障碍[1]。导致血管内皮细胞损伤的因素中,肾素-血管紧张素系统(RAAS),尤其是局部RAAS所产生的血管紧张素Ⅱ(angiotensin ,AngⅡ)在病理生理方面起着重要的作用[2]。异常升高的AngⅡ可通过与血管内皮细胞内的AT1受体结合,引起细胞钙超载并启动一系列的细胞凋亡信息转导过程。替米沙坦(Telmisartan)是新型的长效AngⅡ受体拮抗剂,其作用机制主要是与AT1受体结合,阻断AngⅡ对AT1受体的激动作用,作为临床一线降压药被广泛应用。以往的研究显示,替米沙坦对由高糖或AngⅡ等因素所致的血管内皮细胞损伤具有保护作用[3-6]。静息生理状态下,细胞内钙离子主要储存在内质网内,使细胞内游离钙离子保持在极低的水平约1×10-7mol/L,细胞内外的钙离子浓度差大约为一万倍[7]。当遇到相应的刺激时,细胞会通过多种途径瞬间提高胞内局部或全部的钙离子浓度。其中主要的途径包括胞外钙离子的内流和胞内内质网应激释放储存在其的钙离子。细胞内钙离子浓度在细胞死亡过程中有变化,并且这种变化与许多病理状态相关。已有的研究报道中,在探讨替米沙坦对内皮细胞胞内游离钙离子浓度影响时,较少区分引起胞内钙离子浓度变化的途径或给予限制,本文通过采用无钙离子细胞培养基,消除细胞应激后钙离子从胞外内流的途径,探讨AngⅡ刺激后胞内钙离子的变化,观察替米沙坦对内质网源游离钙离子浓度的调控作用,以细化这两类物质(药物)对胞内钙离子影响及其诱发细胞凋亡的基础研究。
1 材料与方法
1.1 材料
人大动脉血管内皮细胞株(VEC),PriCell公司;替米沙坦,森瑞化工有限公司;Ang Ⅱ,Sigma公司;胎牛血清,美国Hyclone公司;高糖DMEM培养基,美国Hyclone公司;无钙DMEM培养基,美国Hyclone公司;Fluo-3/AM,Sigma公司。替米沙坦和AngⅡ均用无钙无血清DMEM培养液溶解配成10 mg/L和2 μmol/L母液,用时以无钙无血清DMEM细胞培养液调节终末浓度。
1.2 细胞培养
常规复苏血管内皮细胞株,以含血清DMEM培养基(90%高糖DMEM培养基,10%胎牛血清)制备浓度为1×105/mL的血管内皮细胞悬浮液。取10 mL血管内皮细胞悬浮液置于75 cm2无菌培养瓶内,在5%CO2、37℃、湿度95%的条件中孵化培养,隔天置换培养液。当内皮细胞长至80%~90%融合时,以0.25%胰蛋白酶-0.02%EDTA混合液消化传代。当细胞传至第3代时,将细胞接种于35 mm激光共聚焦玻璃底细胞培养皿中,细胞密度为5×104个/mL,置回培养箱中继续培养,待细胞长满后进入分组、Fluo-3/AM负载及干预试验程序。
1.3 实验分组及处理
根据健康人体口服40、80、120 mg替米沙坦片测得的血浆药物峰浓度(Cmax)[8],实验药物浓度设计为60、300、1000 μg/L。
1.3.1 预防性实验 将接种于35 mm细胞培养皿中的血管内皮细胞分为5组:正常对照组、替米沙坦60、300、1000 μg/L预处理组和AngⅡ 0.1 μmol/L组。试验添加药物前每组分别测定初始钙离子浓度(0 h),设为T0。随后,各组培养基中分别加入培养液10 μL(正常对照组)、浓度为0.036 g/L的替米沙坦10 μL(60 μg/L替米沙坦组)、浓度为0.18 g/L的替米沙坦10 μL(300 μg/L替米沙坦组)、浓度0.60 g/L的替米沙坦10 μL(1000 μg/L替米沙坦组)和培养液10 μL(AngⅡ 0.1 μmol/L组)。替米沙坦预处理30 min后检测细胞内游离钙离子浓度(T1),除正常对照组外,其余组均加入浓度2 μmol/L的AngⅡ10 μL,并分别继续共同培养,于AngⅡ加入后瞬时(T2)、0.5 h(T3)、2 h(T4)、8 h(T5)和24 h(T6)抽样检测细胞内游离钙离子浓度。
1.3.2 治疗性实验 将接种于35 mm细胞培养皿中的血管内皮细胞分为5组:正常对照组、AngⅡ 0.1 μmol/L组,AngⅡ+替米沙坦60、300、1000 μg/L组。试验添加药物前每组分别测定初始钙离子浓度(0 h),设为T0。随后,除正常对照组外,其余各组加入 0.1 μmol/L的AngⅡ 10 μL培养30 min后检测细胞内游离钙离子浓度(T1),再加入替米沙坦(加入浓度和体积同“1.3.1”项下),并分别继续共同培养,于替米沙坦加入后瞬时(T2)、0.5 h(T3)、2 h(T4)、8 h(T5)和24 h(T6)抽样检测细胞内游离钙离子浓度。
1.4 胞浆游离钙离子浓度测定[9]
按照检测时间,分别取各组细胞应用激光共聚焦扫描显微镜调节激发波长为488 nm,发射波长为526 nm,置负载Fluo-3的内皮细胞于镜下,于每个检测时点,每组随机选取活细胞8个,各细胞共扫描16次,每次扫描间隔时间为2 s,取16次扫描测得的平均荧光强度数值作为该内皮细胞内钙离子浓度值。
1.5 统计学方法
应用SPSS 13.0统计学软件进行数据分析,计量资料数据用均数±标准差(x±s)表示,多组间比较采用单因素方差分析,组间两两比较采用LSD-t检验,以P < 0.05为差异有统计学意义。
2 结果
2.1 替米沙坦预处理对AngⅡ导致的血管内皮细胞胞内游离钙离子浓度的影响
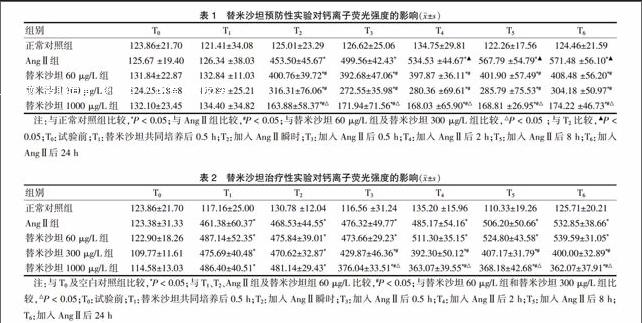
替米沙坦预处理加入AngⅡ 0.1 μmol/L后,胞浆游离钙离子浓度立即(0 h)显著升高,均显著高于正常对照组(P < 0.05),但显著低于AngⅡ组,并且替米沙坦预处理浓度越高抑制作用越强(P < 0.05),作用时间对此无影响。见表1。
2.2 替米沙坦对AngⅡ导致的血管内皮细胞胞内游离钙离子浓度的影响
与正常对照组相比,AngⅡ 0.1 μmol/L诱导30 min,钙离子浓度显著升高(P < 0.05)。与AngⅡ 0.1 μmol/L相比,加入替米沙坦60 μg/L作用24 h,对升高的胞浆游离钙离子浓度无抑制作用;替米沙坦300 μg/L作用2 h或替米沙坦1000 μg/L作用0.5 h才显示其抑制作用(P < 0.05),均显著高于同一时间点的正常对照组;替米沙坦的作用时间对此无影响。见表2。
3 讨论
本试验结果显示,AngⅡ能诱导内质网钙库向胞浆释放游离钙离子,而替米沙坦具有效拮抗AngⅡ对内质网的应激作用及促使胞浆游离钙离子重新被内质网重吸收。血管内皮细胞具有物质转运、自分泌、旁分泌等多重功能,是易受损的功能性界面,对各种不同的病理生理干预均可发生形态学和生物化学方面的改变。多种心血管病变均可引起内源性AngⅡ分泌增加,超生理浓度的AngⅡ可通过与血管内皮细胞内的AT1受体结合,激动G蛋白,使4,5-二磷酸磷脂酰肌醇水解生成三磷酸肌醇(IP3),生成的IP3激动细胞内内质网IP3受体(IP3敏感型钙库),致使内质网向胞浆释放大量游离Ca2+,同时损害内质网钙泵(endoplasmic reticulum Ca2+ ATPase,SERCA)的活力,影响其对细胞质游离钙离子的重新吸收,引起细胞钙超载并启动一系列的细胞凋亡信息转导过程[10-12],这一系列的改变将进一步引起细胞骨架降解,导致细胞出现自溶反应[13-15]。而血管内皮细胞凋亡是动脉粥样硬化病变的独立危险因素[1]。
替米沙坦是新型的AngⅡ受体拮抗剂,其作用机制主要是与AT1受体结合,阻断AngⅡ对AT1受体的激动作用。Selman等[11]对心肌细胞的研究中观察到,替米沙坦能在一定程度上阻断IP3对IP3受体的激动作用,并在进一步的动物试验中观察到,长期给高血压大鼠灌服替米沙坦可令IP3受体的数量下调。Abiko等[10]等Maczewski等[14]研究表明,AngⅡ、甲状腺素等作用内皮细胞后,SERCA mRNA和蛋白质的表达显著下降且和内质网摄取Ca2+的能力下降相关,咪达普利和替米沙坦干预后,SERCA mRNA表达无明显变化,但可显著提高SERCA的活力。
从本试验的预防性和治疗性试验观察到,血管内皮细胞经替米沙坦预处理30 min后均能有效拮抗AngⅡ引起的胞浆游离钙离子浓度上升。这可能与替米沙坦为AngⅡ受体拮抗剂,经替米沙坦预处理30 min后,药物优先占据AngⅡ的AT1受体结合位点,阻碍或(和)减弱了AngⅡ对结合位点的激动作用及由于受体激动后所诱发的连锁反应。替米沙坦的预防作用也可能与其阻断IP3对IP3受体的激动作用,部分阻断钙库内质网钙离子的释放有关。
AngⅡ预先诱导30 min的试验中,300 μg/L替米沙坦作用2 h或1000 μg/L替米沙坦作用0.5 h才显示其抑制胞浆游离钙离子浓度的作用。这可能由于AT1受体被AngⅡ预先占据并激动,而AngⅡ在细胞和组织中的半衰期为15~30 min,在此期间AT1受体等结合位点依然被AngⅡ所占据但由于替米沙坦与AngⅡ竞争相同的作用位点。因此,替米沙坦同样起到一定的拮抗作用,以至于抑制了胞内游离Ca2+的持续上升(与AngⅡ模型组比较)。随着AngⅡ被活细胞逐渐代谢,原先被AngⅡ占据的结合位点重新暴露并被替米沙坦占据。其后胞内游离钙离子浓度的降低可能与沙坦类药物同时抑制多种细胞内氧化反应[16-21],或替米沙坦提高了SERCA的活力,促使胞浆游离钙离子重新被内质网等细胞器重吸收有关。
从观察到的结果可以得出,替米沙坦对AngⅡ引起的胞浆游离钙离子浓度的上升拮抗作用具有剂量相关性,高浓度的替米沙坦拮抗作用明显优于低浓度。但基于受体具有饱和性,组织或细胞中含有某种受体的数目是相对固定的,当受体与配体结合达到饱和时,生物效应并不会因为配体数目的增加而又有增强。故此,替米沙坦或许存在拮抗AngⅡ受体外的其他保护血管内皮细胞的作用机制,如阻断钙离子从内质网中释放、提高SERCA的活力加快胞内游离钙离子的重吸收等,以终止和(或)延缓细胞凋亡的进程。
综上所述,替米沙坦可通过影响AT1受体、IP3受体及SERCA的活力等保护降低内皮细胞内游离钙离子水平,减少细胞钙超载,进而逆转内皮细胞凋亡。
[参考文献]
[1] 方丽娟,刘乃丰.细胞凋亡与动脉粥样硬化关系的研究进展[J].东南大学学报:医学版,2010,29(1):107-110.
[2] 吉亚军,钱春天.替米沙坦对原发性高血压患者血压、左心室重构及血管弹性的影响[J].中国现代医生,2011,49(22):73-75.
[3] 龚丽娅,蓝光明,黄秋霞.替米沙坦改善代谢综合征患者血管内皮功能[J].中国动脉硬化杂志,2011,19(5):431-434.
[4] 龚丽娅,李大强,徐惠兴,等.替米沙坦对代谢综合征患者血管内皮功能的影响[J].海南医学,2011,22(10):35-38.
[5] 李虹,刘强,王宁夫,等.替米沙坦对血管内皮细胞衰老干预作用的实验研究[J].中国临床药学杂志,2009,(6):349-353.
[6] 黄俊,肖静,赫连蔓,秦海燕,等.替米沙坦减轻高糖引起的人血管内皮细胞损伤[J].中华高血压杂志,2008,16(4):314-317.
[7] 陈前,秦环龙.细胞内钙超载与肠上皮细胞凋亡研究[J].中国临床医学,2006,13(1):84-86.
[8] 熊玉卿,李新华,黄鹏,等.替米沙坦在中国健康志愿者体内的药代动力学特征[J].中国药理学通报,2004,20(9):1038-1041.
[9] 苏立,代引,邓武,等.从钙离子调控改变探讨兔肾素血管紧张素系统阻断剂逆转甲状腺素促心肌肥厚的机制[J].中华心血管病杂志,2008,36(8):744-749.
[10] Abiko M,Rodgers KE,Campeau JD,et al. Alterations of angiotensin Ⅱ receptor levels in sutured wounds in rat skin [J]. Invest Sugr,1996,9:447-453.
[11] Selman M,Pardo A. Idio Pathic pulmonary fibrosis:an epithelial/fibroblasticcross-talk disorder [J]. Res Pir Res,2002, 3(1):3.
[12] Kathleen R,Shiquan X,Juna F,et al. DeveloPment of angiotensin(1-7)as anagent to accelerate demral repair [J]. Wound Rep Reg,200,9(3):238-247.
[13] 孟丽,彭瑞云,高亚兵,等.高功率微波辐射后下丘脑神经元凋亡和线粒体膜电位与Ca(2+)的变化[J].中华劳动卫生职业病杂志,2006,24(12):739-741.
[14] Maczewski M,Beresewicz A.Role of nitric oxide and free radicals in cardioprotection by blocking Na+/H+ and Na+/Ca2+ exchange in rat heart [J]. Eur J Pharmaccol,2003,461(2-3):139-147.
[15] Aaronson PI,Robertson TP,Knock GA,et al. Hypoxic pulmonary,vasoconstriction:mechanisms and controversies [J]. J Physiol,2006,570(Pt 1):53-58.
[16] 张浩,张国艳,牛效清.氯沙坦对糖尿病肾病患者氧化应激的影响研究[J].当代医学,2011,17(24):4-6.
[17] 姚丽,魏丹丹,葛丹梅,等.替米沙坦改善维持性血液透析患者微炎症和氧化应激状态[J].中国血液净化,2011, 10(8):426-428.
[18] 姚丽,徐天华,张少青,等.奥美沙坦改善内皮素诱导的大鼠高血压与氧化应激[J].实用药物与临床, 2011,14(3):182-184.
[19] 余莹,李长明,周沛然,等.坎地沙坦在炎症过程中抑制氧化应激反应机制的研究[J].同济大学学报:医学版,2010,31(3):59-63.
[20] 杨宁,付莉,董彦,等. 普罗布考、厄贝沙坦对人脐静脉内皮细胞氧化应激损伤的保护作用及PAI-1含量的影响[J].中国慢性病预防与控制,2010,18(5):505-506.
[21] 唐香,程训民,江时森,等.替米沙坦对1型糖尿病大鼠氧化应激的影响[J].解放军医学杂志,2010,35(7):788-791.
(收稿日期:2014-10-28 本文编辑:程 铭)
猜你喜欢
中国当代医药(2017年5期)2017-04-07
中国当代医药(2017年2期)2017-03-18
中国实用医药(2016年30期)2016-12-28
中国实用医药(2016年23期)2016-12-26
中西医结合心血管病电子杂志(2016年4期)2016-11-30
医学信息(2016年29期)2016-11-28
中国实用医药(2016年25期)2016-11-03
中国实用医药(2016年24期)2016-10-17
中国医药科学(2016年8期)2016-10-09
现代养生·下半月(2015年9期)2015-09-28
