
亚麻木酚素缓释片的质量标准研究
2015-03-09 10:38:00刘效栓毕映燕李季文吴晓琴钱梦茹甘肃省中医院药学部兰州730050
中国药房 2015年9期
刘效栓,毕映燕,李季文,吴晓琴,钱梦茹(甘肃省中医院药学部,兰州 730050)
亚麻木酚素是一种与人体雌激素十分相似的植物雌激素。现代药理研究证实,亚麻木酚素具有抗氧化、延缓衰老[1]、抗肿瘤[2]、预防骨质疏松[3]、降低血清胆固醇[4]、免疫调节等功效,尤其在减轻妇女绝经期症状、预防乳癌和前列腺癌等方面具有良好的疗效[5],且其在预防糖尿病方面也发挥着重要作用[6]。目前,国内外对亚麻木酚素的研究多集中在提取、分离、纯化、药理等方面[7],关于亚麻木酚素类药物制剂的研究则未见文献报道。本研究以亚麻木酚素缓释片为研究对象,采用高效液相色谱(HPLC)法测定其中亚麻木酚素的含量,并对其体外释放度进行研究,以为建立亚麻木酚素缓释片的质量标准、有效控制制剂质量提供参考。
1 材料
1.1 仪器
1525型HPLC仪,包括1525型泵、717型进样器、2487型双通道紫外检测器(美国Waters公司);HS 3120型超声波清洗仪(宁波新基生物科技股份有限公司);BS2245型电子分析天平(德国赛多利斯公司);RZQ-8D型自动溶出取样收集系统(天津市天大天发科技有限公司)。
1.2 药品与试剂
亚麻木酚素缓释片(甘肃省中医院药学部自制,批号:20130306、20130309、20130312);亚麻木酚素对照品(上海同田生物技术有限公司,批号:MUST-12030303,纯度≥98%);甲醇(天津百世化工有限公司,分析纯;天津光复精细化工有限公司,色谱纯);磷酸二氢钾(上海国药集团,分析纯);氢氧化钠(天津市大茂化学试剂厂,分析纯);盐酸(天津百世化工有限公司,分析纯);蒸馏水(实验室自制)。
2 含量测定方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Waters Symmetry Shield-C18(250 mm×4.6 mm,5 μm);流动相:甲醇-水(35∶65,V/V);流速:1.0 ml/min;检测波长:282 nm;柱温:室温;进样量:10 μl。在此色谱条件下,样品中被测成分能够达到基线分离,相邻色谱峰的分离度>1.5,理论板数以亚麻木酚素峰计应大于3 000。
2.2 溶液的制备
2.2.1 对照品溶液 精密称取亚麻木酚素对照品9.0 mg,置于10 ml量瓶中,加甲醇溶解并稀释至刻度,即得。
2.2.2 供试品溶液 取亚麻木酚素缓释片10片,研细,过4号筛,精密称取0.2 g,置于100 ml量瓶中,加甲醇适量,超声(功率:300 W,频率:25 kHz)处理15 min,放凉,加甲醇稀释至刻度,滤过;取续滤液1 ml,置于10 ml量瓶中,加甲醇稀释至刻度,振荡,摇匀,经0.45 μm微孔滤膜滤过,取续滤液,即得。
2.2.3 阴性样品溶液 按亚麻木酚素缓释片的处方工艺,精密称取各辅料适量,按“2.2.2”项下供试品溶液的制备方法制成阴性样品溶液。
2.3 专属性试验
取“2.2”项下对照品、供试品及阴性样品溶液各10 μl,按“2.1”项下色谱条件进样测定,记录色谱,详见图1。结果表明,样品中的辅料成分对测定无干扰。
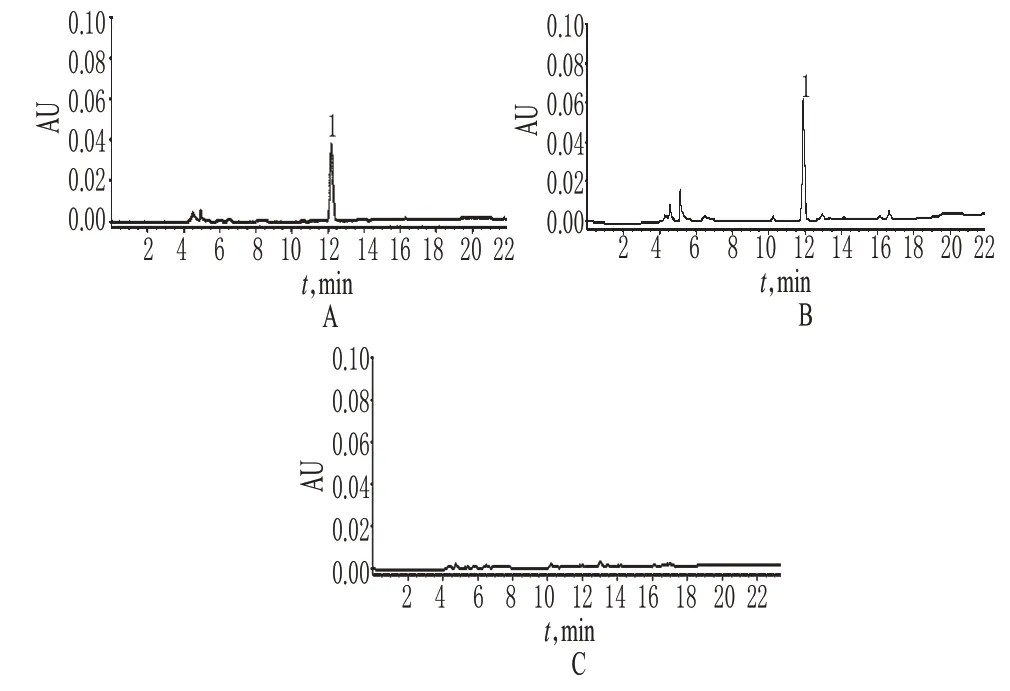
图1 高效液相色谱图A.对照品;B.供试品;C.阴性样品;1.亚麻木酚素Fig 1 HPLC chromatogramsA.substance control;B.test sample;C.negative sample;1.secoisolariciresinol diglucoside
2.4 线性关系考察
精密吸取“2.2.1”项下的亚麻木酚素对照品溶液适量,加甲醇制成每1 ml含亚麻木酚素0.018、0.045、0.090、0.135、0.180、0.225 mg的系列溶液,按“2.1”项下色谱条件进样测定,记录峰面积。以峰面积(y)为纵坐标、亚麻木酚素的质量浓度(x,mg/ml)为横坐标,进行线性回归,得回归方程:y=5×106x+21 041(r=0.999 5)。结果表明,亚麻木酚素的质量浓度在0.018~0.225 mg/ml范围内与峰面积呈良好的线性关系。
2.5 精密度试验
精密吸取“2.2.1”项下的亚麻木酚素对照品溶液10µl,按“2.1”项下色谱条件连续进样6次,记录峰面积。结果,RSD=0.94%(n=6),表明仪器精密度良好。
2.6 稳定性试验
精密吸取同一供试品溶液(批号:20130309)10µl,分别于配制0、4、8、12、18、24 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,RSD=1.99%(n=6),表明供试品溶液在24 h内稳定。
2.7 重复性试验
精密称取同一批亚麻木酚素缓释片(批号:20130309)粉末各适量,共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定。结果,RSD=1.63%(n=6),表明该方法的重复性良好。
2.8 加样回收率试验
精密称取已知含量(150.8 mg/g)的亚麻木酚素缓释片粉末适量,共6份,分别精密加入亚麻木酚素对照品适量,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积,并计算加样回收率,结果见表1。
2.9 样品含量测定
精密称取亚麻木酚素缓释片粉末适量,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积,并计算样品含量,结果见表2。
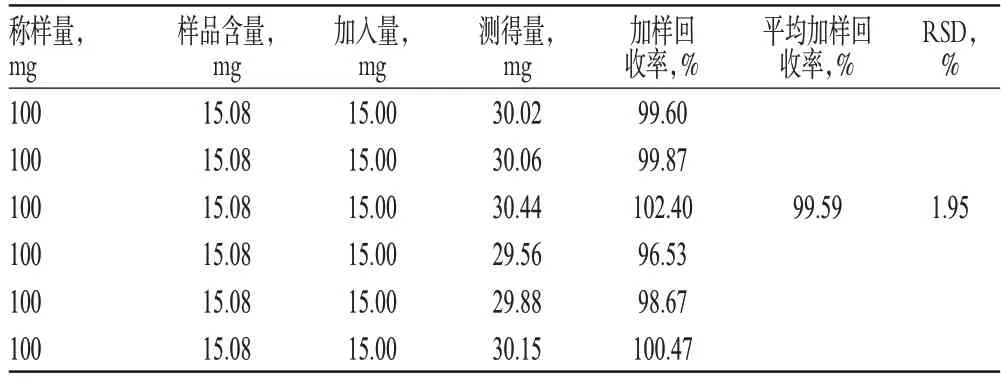
表1 加样回收率试验结果(n=6)Tab 1 Results of recovery tests(n=6)
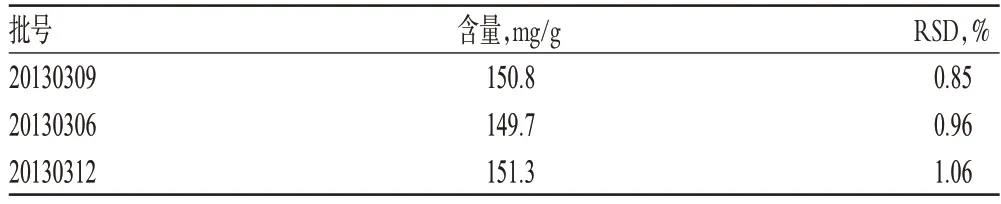
表2 样品含量测定结果(n=3)Tab 2 Results of content determination of samples(n=3)
3 体外释放度测定方法与结果
照2010年版《中国药典》(二部)附录ⅩC溶出度测定法中的转篮法进行。取亚麻木酚素缓释片(批号:20130309)6片,分别置于900 ml蒸馏水中,设定转速100 r/min、温度(37±0.5)℃,在不同取样时间点分别取样5 ml,经0.45 μm微孔滤膜滤过(从取样至滤过应在30 s内完成),同时补充5 ml相同温度、相同体积的水。精密吸取滤液10 μl,按“2.1”项下色谱条件进样测定,记录峰面积,采用外标法计算样品中的药物浓度及每片药物不同时间点的累积释放度,结果见表3。
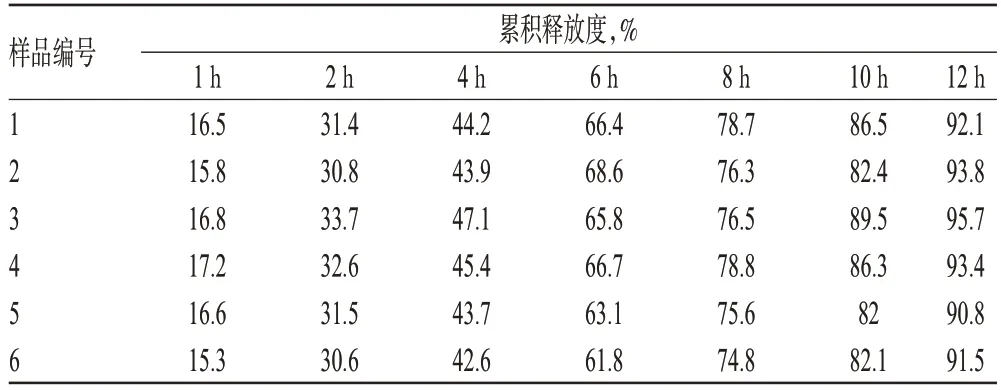
表3 亚麻木酚素缓释片不同时间点的累积释放度(n=6)Tab 3 Results of accumulative releasing ratio at different time points(n=6)
根据2010年版《中国药典》(二部)缓控释制剂指导原则关于取样时间点的规定(第1点的累积释放度约30%,第2点约为50%,最后取样点约为75%),选择取样时间点为2、6、12 h进行累积释放度测定。结果,本品每片在2、6、12 h时的累积释放度分别为10%~30%、50%~70%、80%~100%,均符合规定。
4 讨论
4.1 色谱条件的选择
目前,亚麻木酚素的HPLC法测定多使用梯度洗脱系统[8-9]。笔者曾尝试了不同比例的甲醇-水、乙腈-水、甲醇-酸、乙腈-酸等流动相梯度、等度洗脱系统。结果发现,甲醇、乙腈等度系统均能将亚麻木酚素与相邻杂质峰完全分离,但由于乙腈挥发性较甲醇强,毒性较大,故最后选择甲醇-水等度洗脱作为流动相系统。
4.2 溶出介质的选择
按照释放度测定法[10],笔者考察了水、磷酸盐缓冲液(pH=6.8)、盐酸溶液(0.1 mol/L)对亚麻木酚素释放度的影响。结果发现,3种溶出介质对亚麻木酚素的累积释放度无显著影响,故选择水作为溶出介质。
4.3 体外释放度测定方法的选择
亚麻木酚素缓释片的凝胶骨架材料易吸水膨胀并产生较大的黏性,易粘于杯底,故采用转篮法进行测定。
4.4 检测波长和辅料干扰试验
分别取缓释片样品粉末及其辅料各适量,溶于蒸馏水中,分别在190~390 nm波长范围内进行扫描。结果显示,样品在282 nm波长处有最大吸收,而辅料无吸收,表明辅料对测定无干扰,故最终选择282 nm作为检测波长。
综上所述,该方法准确、简便,专属性、重复性好,可用于亚麻木酚素缓释片的质量控制。
[1]房娜.亚麻木酚素抗氧化产品开发[D].济南:山东师范大学,2013.
[2]McCann SE,Muti P,Vito D,et al.Dietary lignan intakes and risk of pre-andpostmenopausal breast cancer[J].Int J Cancer,2004,111(3):440.
[3]那晓琳,赵新宇,李丽娜,等.亚麻木酚素对去卵巢大鼠血脂水平的影响[J].营养学报,2012,34(2):159.
[4]张进丽.亚麻木酚素(SDG)治疗中老年女性骨质疏松症的临床研究[J].中国现代医学杂志,2013,23(31):65.
[5]张文斌,许时婴.亚麻木酚素的微波辅助提取工艺研究[J].天然产物研究与开发,2006,18(2):286.
[6]沈晓东,李多伟,赵蓉,等.亚麻木酚素的研究进展[J].中成药,2009,31(4):598.
[7]张文斌,许时婴.大孔吸附树脂法分离纯化亚麻木酚素的研究[J].食品工业科技,2006,27(10):96.
[8]Jatinder KM,Valeriya KR,Shankar PS,et al.HPLC method with fluorescence detection for the quantitative determination of flaxseed lignans[J].J of Chromatography B,2010,878(30):3 076.
[9]龙惊惊.亚麻籽中亚麻油和木酚素SDG的提取工艺研究[D].哈尔滨:东北林业大学,2012.
[10]国家药典委员会.中华人民共和国药典:一部[S].2010年版.北京:中国医药科技出版社,2010:附录87.
猜你喜欢
中华诗词(2022年6期)2022-12-31 06:42:40
纺织服装流行趋势展望(2020年1期)2020-02-01 06:33:14
当代陕西(2019年16期)2019-09-25 07:28:42
中成药(2018年6期)2018-07-11 03:01:10
鹿鸣(2018年1期)2018-01-30 12:20:08
中成药(2017年5期)2017-06-13 13:01:12
南方文学(2016年3期)2016-06-12 13:54:34
纺织服装流行趋势展望(2016年6期)2016-05-04 03:53:28
纺织服装流行趋势展望(2016年4期)2016-05-04 03:50:43
纺织服装流行趋势展望(2016年1期)2016-05-04 03:46:02
