
续骨冲和膏的质量标准研究
2015-03-12 08:56吴凤荣曾聪彦胡玉良卢建业广州中医药大学附属中山中医院广东中山5840广东药学院中药学院广东中山58400
中国药房 2015年9期
吴凤荣,曾聪彦,胡玉良,卢建业(.广州中医药大学附属中山中医院,广东中山 5840;.广东药学院中药学院,广东中山 58400)
续骨冲和膏由大黄、赤芍、当归、续断、黄柏、独活等20味中药制备而成,是广州中医药大学附属中山中医院的常用经验方,具有活血通络、消肿止痛、疗伤续骨的功效,主要用于治疗跌打瘀伤。由于该制剂尚未建立完善的质量标准,为了控制其产品质量,保证其安全性和有效性,本研究采用薄层色谱(TLC)法对该制剂中的大黄、黄柏、赤芍、当归、骨碎补、独活、荆芥、木香等多味药进行了定性鉴别,并采用高效液相色谱(HPLC)法对制剂中的君药大黄所含的大黄素和大黄酚进行了含量测定。
1 材料
1.1 仪器
1100型HPLC仪(美国安捷伦公司);KQ3200E型医用超声波清洗器(昆山市超声仪器有限公司);BS224S型电子天平(德国赛多利斯公司);电热恒温水浴锅(上海衡平仪器仪表厂);TC-15型套式恒温器(浙江新华医疗器械厂)。
1.2 药品与试剂
续骨冲和膏(批号:20131109、20131202、20140108)由广东省中山市中医院制剂室提供;大黄素(批号:110756-200110)、大黄酚(批号:110796-201118)、芍药苷(批号:110736-201136)对照品及黄柏(批号:121510-201105)、大黄(批号:121249-201003)、赤芍(批号:121093-200402)、当归(批号:120927-201315)、骨碎补(批号:121169-200503)、独活(批号:120940-201111)、木香(批号:120921-201008)、荆芥(批号:120911-201110)对照药材均购自中国食品药品检定研究院;硅胶G(青岛海洋化工有限公司分厂,批号:20120208);其余试剂均为市售分析纯。
2 方法与结果
2.1 TLC鉴别
2.1.1 大黄[1]取本品1.5 g,用玻璃棒分散,加甲醇20 ml,浸泡1 h,滤过,取滤液5 ml,蒸干,残渣加水10 ml使溶解,加盐酸1 ml,水浴加热回流30 min,立即冷却,用乙醚分2次振摇提取,每次20 ml,合并乙醚液,蒸干,残渣加三氯甲烷1 ml使溶解,作为供试品溶液。取大黄对照药材0.1 g,加甲醇20 ml,按供试品溶液的制备方法制成对照药材溶液。取缺大黄的其余药味适量,按处方工艺制成缺大黄的阴性对照品,取约1.5 g,按上述供试品溶液的制备方法制成缺大黄的阴性对照溶液。照TLC法[2010年版《中国药典》(一部)附录ⅥB]试验,吸取上述3种溶液各5 μl分别点于同一硅胶G薄层板上,以石油醚(30~60 ℃)-甲酸乙酯-甲酸(15∶5∶1,V/V/V)的上层溶液为展开剂,展开,取出,晾干,分别置于日光和紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照药材色谱相应的位置上,显相同的橙黄色荧光斑点;置氨蒸气中熏后,斑点变为红色。大黄的TLC图见图1。
2.1.2 黄柏[2-3]取本品3 g,用玻璃棒分散,加1%醋酸甲醇溶液20 ml,超声(功率:150 W,频率:40 kHz)处理20 min,滤过,滤液浓缩至1 ml,作为供试品溶液。取黄柏对照药材0.1 g,加1%醋酸甲醇20 ml,按供试品溶液的制备方法制成对照药材溶液。取缺黄柏的其余药味制成的阴性对照品3 g,按上述供试品溶液的制备方法制成缺黄柏的阴性对照溶液。照TLC法[2010年版《中国药典》(一部)附录ⅥB]试验,吸取供试品溶液、阴性对照溶液各5 μl及对照药材溶液1 μl分别点于同一硅胶G薄层板上,以乙酸乙酯-丁酮-甲酸-水(10∶6∶1∶1,V/V/V/V)为展开剂,展开,取出,晾干,分别置日光和紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的黄色斑点和荧光斑点。黄柏的TLC图见图2。

图1 大黄的TLC图A.熏氨蒸气前;B.熏氨蒸气后;C.紫外光灯下;1~3.供试品;4.大黄对照药材;5缺大黄的阴性样品Fig 1 TLC of Rhei RadixA.before fumed with ammonia vapor;B.after fumed with ammonia vapor;C.under UV lamp;1-3.test samples;4.Rhei Radix reference substance;5.negative control without Rhei Radix
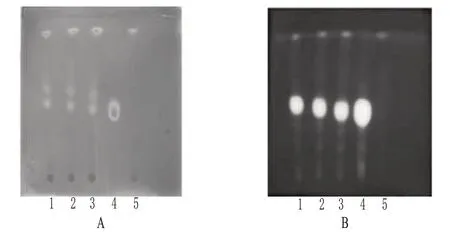
图2 黄柏的TLC图A.日光下;B.紫外光灯下;1~3.供试品;4.黄柏对照药材;5.缺黄柏的阴性样品Fig 2 TLC ofP.chineseA.under sunlight;B.under UV lamp;1-3.test samples;4.Phellodendri Chinensis reference substance;5.negative control withoutP.chinese
2.1.3 赤芍[4]取本品22 g,用玻璃棒分散,加乙醇50 ml,振摇5 min,滤过,滤液蒸干,残渣加乙醇2 ml使溶解,作为供试品溶液。取赤芍对照药材0.5 g,加乙醇10 ml,按上述供试品溶液的制备方法制成对照药材溶液。取缺赤芍的其余药味制成的阴性对照品22 g,按上述供试品溶液的制备方法制成缺赤芍的阴性对照溶液。照TLC法[2010年版《中国药典》(一部)附录ⅥB]试验,吸取上述3种溶液各5 μl分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(40∶5∶10∶0.2,V/V/V/V)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,加热至斑点显色清晰。结果,供试品色谱中,在与对照药材色谱相应的位置上,显相同的蓝紫色荧光斑点。赤芍的TLC图见图3。
2.1.4 当归[5]取本品22 g,加乙醚50 ml,超声(功率:150 W,频率:40 kHz)处理10 min,滤液蒸干,残渣加乙醇1 ml使溶解,作为供试品溶液。取当归对照药材0.5 g,加乙醚20 ml,按上述供试品溶液的制备方法制成对照药材溶液。取缺当归的其余药味制成的阴性对照品22 g,按上述供试品溶液的制备方法制成缺当归的阴性对照溶液。照TLC法[2010年版《中国药典》(一部)附录ⅥB]试验,吸取上述3种溶液各10 μl分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯(4∶1,V/V)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。当归的TLC图见图4。
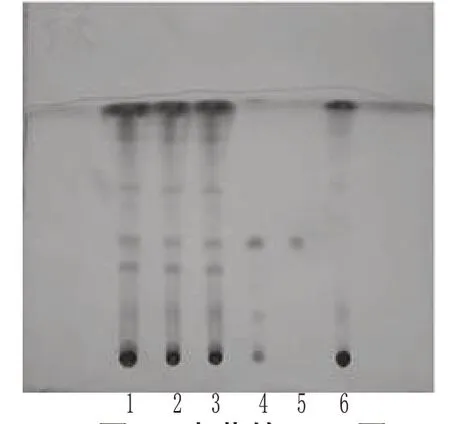
图3 赤芍的TLC图1~3.供试品;4.赤芍对照药材;5.芍药苷对照品;6.缺赤芍的阴性样品Fig 3 TLC of Paeoniae Radix Rubra1-3.test samples;4.Paeoniae Radix Rubra reference substance;5.peoniflorin control;6.negative control without Paeoniae Radix Rubra

图4 当归的TLC图1~3.供试品;4.当归对照药材;5.缺当归的阴性对照品Fig 4 TLC ofA.sinensis1-3.test samples;4.Angelicae Sinensis Radix reference substance;5.negative control withoutA.sinensis
2.1.5 木香[6]取本品20 g,加甲醇40 ml,用玻璃棒分散,超声(功率:150 W,频率:40 kHz)处理30 min,滤过,取滤液浓缩至2 ml,作为供试品溶液。取木香对照药材0.5 g,加甲醇10 ml,按供试品溶液的制备方法制成对照药材溶液。取缺木香的其余药味制成的阴性对照品0.5 g,按上述供试品溶液的制备方法制成缺木香的阴性对照溶液。照TLC法[2010年版《中国药典》(一部)附录ⅥB]试验,吸取上述3种溶液各5 μl分别点于同一硅胶G薄层板上,以三氯甲烷-环己烷-甲醇(5∶3∶0.1,V/V/V)为展开剂,展开,取出,晾干,喷以1%香草醛硫酸溶液,加热至斑点显色清晰。结果,供试品色谱中,在与对照药材色谱相应的位置上显相同颜色的荧光斑点。木香的TLC图见图5。
2.2 大黄素和大黄酚的含量测定[7-8]
2.2.1 色谱条件与系统适用性试验 色谱柱:Boston Green ODS C18(250 mm × 4.6 mm,5 μm);流动相:甲醇-0.1%磷酸溶液(85∶15,V/V);柱温:25 ℃;检测波长:254 nm。理论板数按大黄素、大黄酚峰计应不低于3 000。
2.2.2 供试品溶液的制备[1]精密称取本品约3 g,置于100 ml锥形瓶中,精密加入甲醇25 ml,称定质量,加热回流提取30 min,放冷,再次精密称定,加甲醇补足减失的质量,摇匀,滤过。精密量取续滤液10 ml,置圆底烧瓶中,挥干溶剂,加水15 ml使溶解,再加盐酸1 ml,超声(功率:150 W,频率:40 kHz)处理5 min,加热回流提取30 min,立即冷却,置于分液漏斗中。用少量乙醚洗涤容器,洗涤液并入分液漏斗中,用乙醚分3次振摇提取,每次15 ml,合并乙醚液,蒸干。残渣加甲醇使溶解,转移至25 ml量瓶中,加甲醇定容,摇匀,经0.45 μm微孔滤膜滤过,取续滤液,即得。
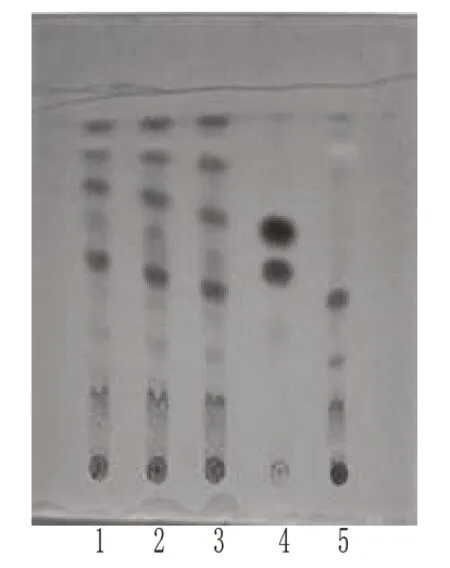
图5 木香的TLC图1~3.供试品;4.木香对照药材;5.缺木香的阴性对照品Fig 5 TLC ofA.lappa1-3.test samples;4.Aucklandiae Radix reference substance;5.negative control withoutA.lappa
2.2.3 混合对照品溶液的制备 精密称取大黄素、大黄酚对照品各适量,加甲醇制成每1 ml含48.25 μg大黄素和109 μg大黄酚的混合对照品溶液。
2.2.4 阴性样品溶液的制备 按处方比例称取药材,按续骨冲和膏的处方工艺制成缺大黄的阴性样品,再按“2.2.2”项下方法制成缺大黄的阴性样品溶液。
2.2.5 专属性试验 分别精密吸取混合对照品溶液、供试品溶液及阴性样品溶液各20 μl,按“2.2.1”项下色谱条件进样测定。结果,供试品色谱中,在与对照品色谱相应保留时间处,有相同的色谱峰;阴性对照无干扰。色谱见图6。
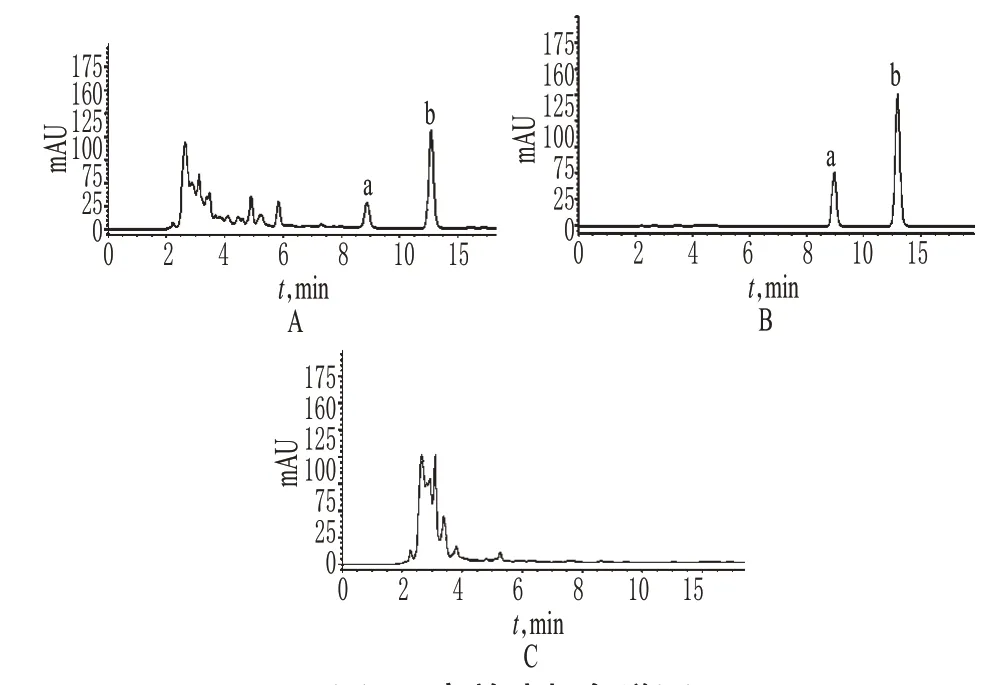
图6 高效液相色谱图A.供试品;B.混合对照品;C.阴性对照;a.大黄素;b.大黄酚Fig 6 HPLC chromatogramsA.test sample;B.mixed control;C.negative control;a.emodin;b.chrysophanol
2.2.6 线性关系考察 精密量取“2.2.3”项下的混合对照品溶液0.5、1、2、4、8 ml,分别用甲醇稀释,得到系列浓度的混合对照品溶液。分别精密吸取系列对照品溶液各20 μl,按“2.2.1”项下色谱条件进样测定,记录峰面积。以质量浓度(x,μg)为横坐标、峰面积(y)为纵坐标,绘制标准曲线,得大黄素的回归方程为y=72.876x-30.738(r=0.999 9),大黄酚的回归方程为y=62.82x-57.904(r=0.999 9)。结果表明,大黄素和大黄酚的质量浓度分别在2.41~38.6、5.45~87.2 μg/ml范围内与各自峰面积呈良好线性关系。
2.2.7 精密度试验 精密称取续骨冲和膏(批号:201301202)样品适量,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件连续进样5次,记录峰面积。结果,大黄素和大黄酚峰面积的RSD分别为1.3%、1.2%(n=5),表明仪器精密度良好。
2.2.8 稳定性试验 精密吸取同一批供试品溶液各适量,分别于配制0、2、4、6、8 h时按“2.2.1”项下色谱条件进样测定,记录峰面积。结果,大黄素和大黄酚峰面积的RSD分别为1.08%、1.53%(n=5),表明供试品溶液在8 h内稳定性良好。
2.2.9 重复性试验 称取同一批次的续骨冲和膏(批号:201301202)共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积,计算样品含量。结果,大黄素和大黄酚的平均含量分别为0.15、0.33 mg/g,其峰面积的RSD分别为1.2%、1.1%(n=6),表明该方法重复性良好。
2.2.10 加样回收率试验 精密称取同一批次(批号:201301202)已知含量的续骨冲和膏1 g(约含大黄素0.15 mg、大黄酚0.33 mg),共6份,分别置于磨口锥形瓶中,精密加入混合对照品溶液适量,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积,并计算加样回收率,结果见表1。
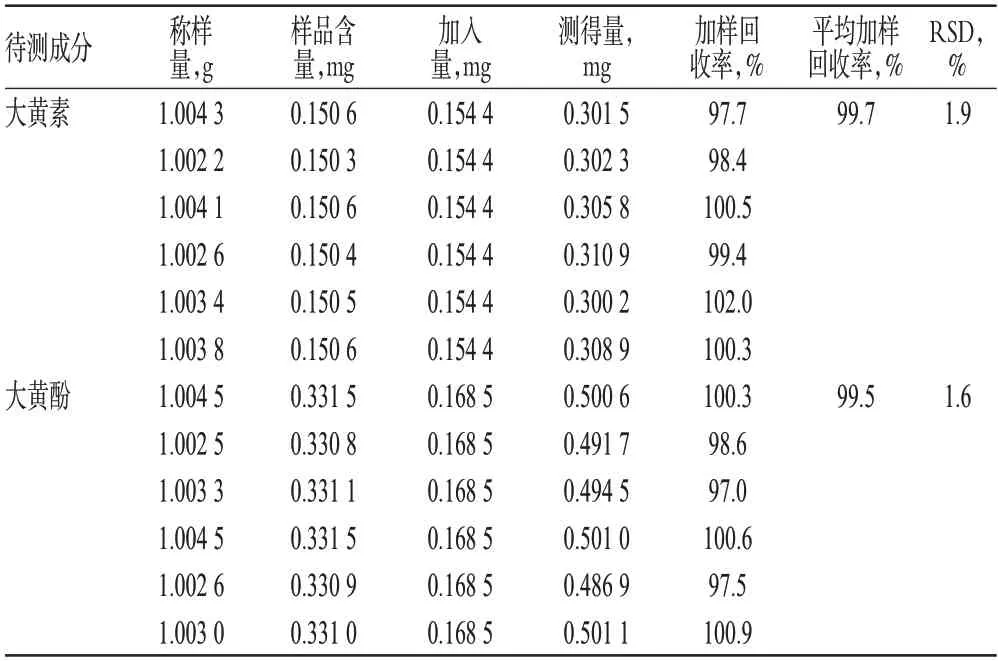
表1 加样回收率试验结果(n=6)Tab 1 Results of recovery tests(n=6)
2.2.11 样品含量测定 精密称取3批样品各适量,分别按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积,并计算样品含量,结果见表2。
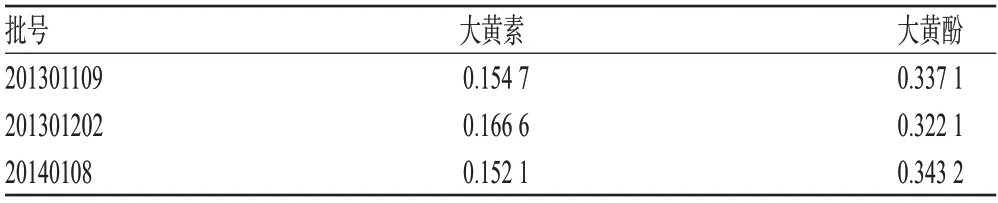
表2 样品含量测定结果(mg/g,n=3)Tab 2 Results of content determination(mg/g,n=3)
3 讨论
本制剂中各药味的TLC鉴别试验基本参照2010年版《中国药典》(一部)中有关药味的鉴别方法。其中,大黄、赤芍、当归的TLC鉴别试验方法分别与2010年版《中国药典》方法基本一致。黄柏原采用2010年版《中国药典》中的鉴别条件,以三氯甲烷-甲醇-水(30∶15∶4,V/V/V)的下层溶液为展开剂[2],结果斑点不清晰;后尝试采用文献方法[3]的乙酸乙酯-丁酮-甲酸-水(10∶6∶1∶1,V/V/V/V)为展开剂,结果斑点清晰,分离度好。木香原采用2010年版《中国药典》中的鉴别条件,以环己烷-甲酸乙酯-甲酸(15∶5∶1,V/V/V)的上层溶液为展开剂[2],结果展开效果也很差,后参考文献方法[4]改成三氯甲烷-环己烷-甲醇为展开剂,且经不断调整各试剂比例,直至调整到5∶3∶0.1(V/V/V)时,其展开效果好。由于本制剂是由20味中药组成的复方制剂,成分较为复杂,TLC鉴别过程中对提取方法及展开剂的选择有较高要求,在对处方中骨碎补、续断、防风、红花、蒲黄、独活等其余药味的TLC鉴别研究中,存在图谱斑点不清晰、拖尾、阴性干扰大等缺陷,需进一步研究,暂不列入标准中。
采用HPLC法测定大黄素和大黄酚含量时,供试品溶液的制备方法则参照了有关文献方法,简化了2010年版《中国药典》中大黄含量测定项[2]中配制8%盐酸溶液的步骤,缩短了回流提取时间。参照2010年版《中国药典》(一部)中大黄含量测定项下的色谱条件,以甲醇-0.1%磷酸溶液(75∶25、80∶20、85∶15,V/V)多种比例试验选择流动相,当比例为85∶15时,大黄素和大黄酚的色谱峰分离度适合、基线平直、峰形对称、其余成分无干扰。
综上所述,本研究所建立的方法方便快捷,重现性、专属性好,结果稳定可靠,可作为续骨冲和膏的质量控制方法。
[1]刘起华,张萱,文谨.高效液相色谱法测定四黄膏中大黄素、大黄酚的含量[J].中国现代应用药学杂志,2007,24(1):6.
[2]国家药典委员会.中华人民共和国药典:一部[S].2010年版.北京:中国医药科技出版社,2010:286、58、22.
[3]王丽军,刘丽萍.黄柏止痒洗剂中黄柏、苦参的薄层色谱鉴别[J].陕西中医,2011,32(10):1 407.
[4]李伯群,彭腾,余蕾,等.祛毒胶囊质量标准研究[J].中国药房,2007,18(12):925.
[5]黄艳萍,黄勇红.当归调经片的薄层鉴别和阿魏酸的测定[J].中国实验方剂学杂志,2010,16(6):119.
[6]齐双山,王青虎,包巴特尔,等.蒙成药中木香的薄层色谱鉴别研究[J].时珍国医国药,2001,12(5):430.
[7]赵嘉将,刘静,龚元香,等.沉香化气片的薄层色谱鉴别[J].中国药业,2008,17(20):39.
[8]曹文静,郭姗姗,笔雪梅.HPLC测定结核丸中大黄的5种有效成分含量[J].黑龙江医药,2014,27(4):733.
猜你喜欢
中草药(2022年16期)2022-08-16
科学家(2022年3期)2022-04-11
海峡姐妹(2019年8期)2019-09-03
青年歌声(2019年4期)2019-04-11
天然产物研究与开发(2019年1期)2019-03-01
天然产物研究与开发(2018年6期)2018-07-09
天然产物研究与开发(2018年1期)2018-02-02
中成药(2017年12期)2018-01-19
中成药(2017年4期)2017-05-17
人民周刊(2016年11期)2016-06-30
