
Differential mRNA expression profiling of oral squamous cell carcinoma by high-throughput RNA sequencing
2015-02-14 02:40:08LiangyuGeSiyuLiuLongXieLeiSangChangyanMaHongweiLi
Liangyu Ge,Siyu Liu,Long Xie,Lei Sang,Changyan Ma,Hongwei Li,✉
Differential mRNA expression profiling of oral squamous cell carcinoma by high-throughput RNA sequencing
Liangyu Ge1,2,△,Siyu Liu1,2,△,Long Xie1,2,Lei Sang3,Changyan Ma4,Hongwei Li1,2,✉
1Jiangsu Key Laboratory of Oral Diseases,Nanjing Medical University,Nanjing,Jiangsu 210029,China.
2Department of Oral and Maxillofacial Surgery,Affiliated Hospital of Stomatology,Nanjing Medical University,Nanjing, Jiangsu 210029,China;
3Department of Oral and Maxillofacial Surgery,Suzhou Huaxia Stomatological Hospital,Suzhou,Jiangsu 215002,China;
4Department of Developmental Genetics,Nanjing Medical University,Nanjing,Jiangsu 210029,China.
Differentially expressed genes are thought to regulate the development and progression of oral squamous cell carcinomas(OSCC).The purpose of this study was to screen differentially expressed mRNAs in OSCC and matched paraneoplastic normal tissues,and to explore the intrinsic mechanism of OSCC development and progression.We obtained the differentially expressed mRNA expression profiles in 10 pairs of fresh-frozen OSCC tissue specimens and matched paraneoplastic normal tissue specimens by high-throughput RNA sequencing.By using Gene Ontology enrichment analysis and Kyoto Encyclopedia of Genes and Genomes(KEGG)pathway enrichment analyses,the functional significance of the differentially expressed genes were analyzed.We identified 1,120 significantly up-regulated mRNAs and 178 significantly down-regulated mRNAs in OSCC,compared to normal tissue. The differentially expressed mRNAs were involved in 20 biological processes and 68 signal pathways.Compared to adjacent normal tissue,the expression of MAGEA11 was up-regulated;TCHH was down-regulated.These findings were verified by real-time PCR.These differentially expressed mRNAs may function as oncogenes or tumor suppressors in the development and progression of OSCC.This study provides novel insights into OSCC.However, further work is needed to determine if these differentially expressed mRNAs have potential roles as diagnostic biomarkers and candidate therapeutic targets for OSCC.
oral squamous cell carcinoma,high-throughput RNA sequencing,mRNA,Gene Ontology,KEGG pathway
Introduction
Oral squamous cell carcinoma(OSCC)is the sixth most common cancer worldwide.It is the cause of over 400,000 cancer related deaths each year,with 80% of deaths occurring in developing countries[1].The incidence rate of OSCC is increasing,especially in younger people[2].In addition,OSCC accounts for over 40% of head and neck squamous cell carcinoma(HNSCC). According to the American Cancer Society,while the overall cancer cases have increased by approximately 8% in the last five years,the number of new cases ofOSCC has increased by approximately 21%[3].OSCC has a very poor prognosis due to its invasive nature, and despite advances in treatment and diagnostics,the five-year survival rate is below 50% and has not improved in the last three decades[2].
In common with other cancers,the development and progression of OSCC is a multistep process with the accumulation of genetic and epigenetic changes[4].It is generally accepted that many of cancer-associated gene changes originate with the gain,loss,or mutation of genetic information[5].Therefore,studying these genetic changes improves understanding of the molecular basis of cancer in order to deliver potential diagnostic or outcome markers,and therapeutic targets for cancer patients.
Recent studies have indicated that changes of mRNA expression levels in OSCC is associated with tumor development,maintenance,and progression[6-7]. As a consequence,inducing or silencing mRNA expression of these oncogenes or anti-oncogenes has been used as a method to control and treat neoplasms[8-9].In addition,mRNAs could potentially be used as biomarkers of OSCC or other oral cancers[10-11].
In large-scale clinical studies,genome-wide gene expression profiling can advance the discovery of cancer biomarkers and therapeutic targets.The human transcriptome is highly complex;the human genome encodes 20,000-25,000 genes.In contrast,because each gene can produce multiple transcripts and,thereby,multiple proteins,the proteome is many times larger and diverse in its functions[12].Therefore,efficient sequencing technologies are urgently required.
High-throughput RNA sequencing(RNA-Seq)and the rapid development of new technologies is providing unprecedented opportunities for interrogating the human transcriptome[13].This has led to next generation sequencing(NGS)applications,such as Illumina/Solexa, Applied Biosystems/SOLiD,and Roche/454 FLX[14]. Compared to traditional techniques,NGS offers greatly improved dynamic ranges and specificity for transcriptome analyses,while sample throughput is continuously increasing and costs are being reduced[15].
Although there have been published reports on the successful construction of genome-wide mRNA expression profiles in other types of cancer,highlighting the power and capabilities of high-throughput sequencing techniques,there have been relatively few relating to OSCC[16-17].In this study,we exploited high-throughput RNA-Seq technology to construct comprehensive mRNA expression profiles of OSCC and matched paraneoplastic normal tissues.We used the information to explore and predict the molecular functions and biological pathways of the differentially expressed mRNAs.Our results have provided a basis for identifying further molecular markers for the diagnosis and treatment of OSCC.
Materials and methods
Tissue samples
All the samples used in this study were selected from OSCC patients treated at Affiliated Stomatological Hospital of Nanjing Medical University between November 2011 and February 2012.Written informed consent was obtained from all patients and the study protocol was approved by the Institutional Review Board of Affiliated Stomatological Hospital of Nanjing Medical University.Ten pairs of OSCC tissue and matched para-neoplastic normal tissue specimens,which had been identified by pathologists,were collected and fresh-frozen immediately after surgery,and stored at-80°C. The proportion of cancer cells in a tissue section was greater than 80%.None of the patients had received preoperative chemotherapy,radiotherapy,or any other anticancer treatment prior to surgery.Clinical characteristics of the patients are given inTable 1.

Table 1 Clinicopathologic data of 10 patients included in the study
Illumina Solexa sequencing
Total RNA was isolated from the 10 paired fresh-frozen OSCC tissue and matched paraneoplastic normal tissue specimens using TRIzol®(Invitrogen,Carlsbad, CA,USA)according to the manufacturer's protocol.The integrity of the RNA was evaluated using the Agilent2100 BioAnalyzer(Agilent Technologies,Palo Alto,CA, US).The mRNA was purified from extracted total RNA using Oligo(dT)magnetic beads adsorption,and converted into double-stranded cDNA by reverse transcription.Nla III was used to digest the cDNA and the Illumina adaptor 1 was ligated.Mme I was used to digest the CATG site and the Illumina adaptor 2 was ligated to the 3′end.Primer GX1 and GX2 were used for PCR. The DNA was purified,and sequencing was performed by the Illumina Cluster Station and Genome Analyzer (Illumina Inc.,CA,USA)at Beijing Genomics Institute, Shenzhen,according to the manufacturer's protocol.
Data processing
Sequencing-received raw image data were transformed by base calling into sequence data(termed raw data or raw reads).Dirty tags were filtered through data processing steps.Clean tags were obtained by sequence quality assessment;saturation analysis of sequencing,and experimental repeatability analysis.Subsequently,all clean tags were mapped to the reference sequences and only one-bp mismatch was accepted.Clean tags mapped to reference sequences from multiple genes were filtered and the remaining clean tags were designated as unambiguous clean tags.The number of unambiguous clean tags for each gene was calculated and normalized to the number of transcripts per million(TPM) of clean tags[18-19].
Screening of differentially expressed genes
Differentially expressed genes(DEGs)between OSCC samples and their matched paraneoplastic normal samples were identified by a rigorous algorithm[20].False discovery rate(FDR)was used as a statistical method to determine the threshold P-value by multiple testing and analyzed by manipulating the FDR value[21].The levels of mRNA expressions in the paired samples were calculated by log2Ratio.For this study,we applied an FDR≤0.001 and the absolute value of log2Ratio≥1 as the threshold to judge the significance of gene expression difference.
Gene Ontology functional enrichment analysis for DEGs
For gene expression profiling analysis,Gene Ontology (GO)enrichment analyses of functional significance were performed by hypergeometric testing to map all differentially expressed genes to GO terms(each GO term belongs to a type of ontology in the GO database; http://www.geneontology.org).Searches were carried out for significantly enriched GO terms in DEGs,compared to the genome background.The P-value was corrected by Bonferroni and a corrected P-value of 0.05 was used as the threshold to judge the differential gene expression of significantly enriched GO terms.
Pathway enrichment analysis of DEGs
The Kyoto Encyclopedia of Genes and Genomes (KEGG;http://www.genome.jp/kegg)[22]was used for pathway enrichment analysis of significance by using the hypergeometric test to map all DEGs and search for pathways significantly enriched in DEGs,compared to the genome background.We defined a Q-value of 0.05 as the threshold to judge the differential gene expression of significantly enriched pathways.
Real-time PCR
We selected one of the most up-regulated mRNAs (MAGEA11)and one of the most down-regulated mRNAs(TCHH)for confirmation against 15 other paired OSCC tissue and adjacent normal tissue samples. SnRNA U6 was used as the endogenous control.Specific primer sets for MAGEA11,TCHH and U6 were obtained from Genecopoeia,Guangzhou,China.
For each mRNA amplification,a total reaction volume of 20 μL containing 2×SYBR Green Real-time PCR master mix,mRNA primers,the cDNA template,and diethyl pyrocarbonate(DEPC)-treated water was used. The PCR procedure was as follows:95°C for 5 minutes; 40×15 seconds cycles at 95°C;60°C for 30 seconds; and 72°C for 30 seconds.To quantify changes in gene expression,relative mRNA expressions were calculated according to the formula;2-ΔΔCt[19].
Statistical analysis
Data was expressed as mean±SEM.Statistical analysis was performed using SPSS v17.0 software and Student’s t-test was used to analyze the OSCC tissue and adjacent normal tissue samples.P<0.05 was considered statistically significant.
Results
Differentially expressed mRNAs
By constructing the mRNA expression profile of OSCC and analyzing the sequence data,we identified 1,120 significantly up-regulated mRNAs and 178 significantly down-regulated mRNAs.These includedMAGEA11(log2Ratio=10.252665),which was up-regulated>1,000-fold,and TCHH(log2Ratio= -10.759888),which was down-regulated>1,700-fold. The 10 most up-regulated and down-regulated mRNAs are shown inTable 2.
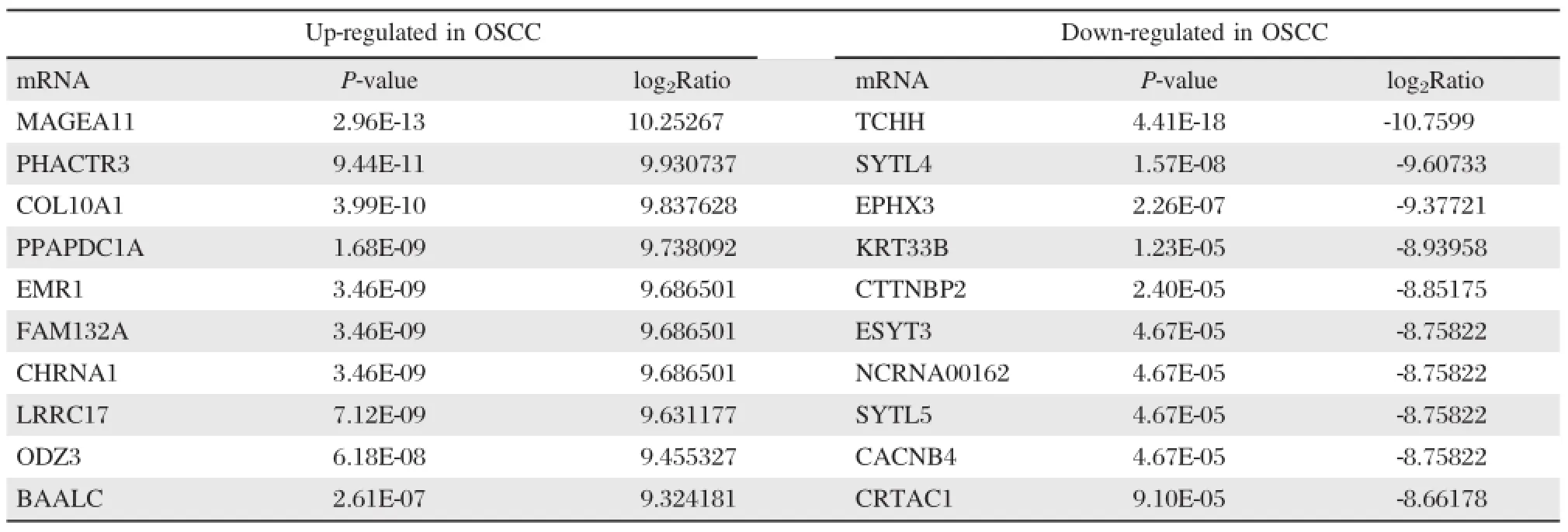
Table 2 The 10 most up-regulated and down-regulated mRNAs in OSCC
GO functional enrichment analysis:cellular components
Through GO enrichment analysis,we determined that the differentially expressed mRNAs were significantly enriched in the following cellular components:extracellular region;extracellular matrix;proteinaceous extracellular matrix;collagen;cytoplasm;endoplasmic reticulum; macromolecular compounds;fiber collagen(Table 3).
(1)规范护理文件书写:组织全体护理人员学习《护理病历书写要求及质量标准》,规范护理文件书写,确保护理记录可作为有力的法律证据之一。(2)进一步学习理论、技能及操作规范:随着新技术的开展和新药品的应用,护理人员应该不断地学习与实践[5]。
GO functional enrichment analysis:molecular functions
The results of GO molecular function analysis showed that the differentially expressed mRNAs were significantly enriched in the following molecular functions: endopeptidase activity;growth factor binding;peptidase activity;protein binding,peptidase activity,acting on L-amino acid peptides;oxidoreductase activity;and cytokine activity(Table 4).Protein binding was found to be the function with the highest level of enrichment with 353 differentially expressed mRNAs.
To validate the results of deep sequencing,the differential expression of MAGEA11 and TCHH were verified by real-time PCR against 15 paired OSCC and adjacent normal tissues.The results showed that MAGEA11 expression was up-regulated in the tumor samples.The mean expression of MAGEA11 in OSCC tissues(3.0518±1.5700) was significantly higher than in normal tissues (P<0.05;Fig.1A);TCHH expression was down-regulated in the tumor samples.The mean expression of TCHH in OSCC tissues(0.5388±0.1700)was significantly lower than normal tissues(P<0.05;Fig.1B).
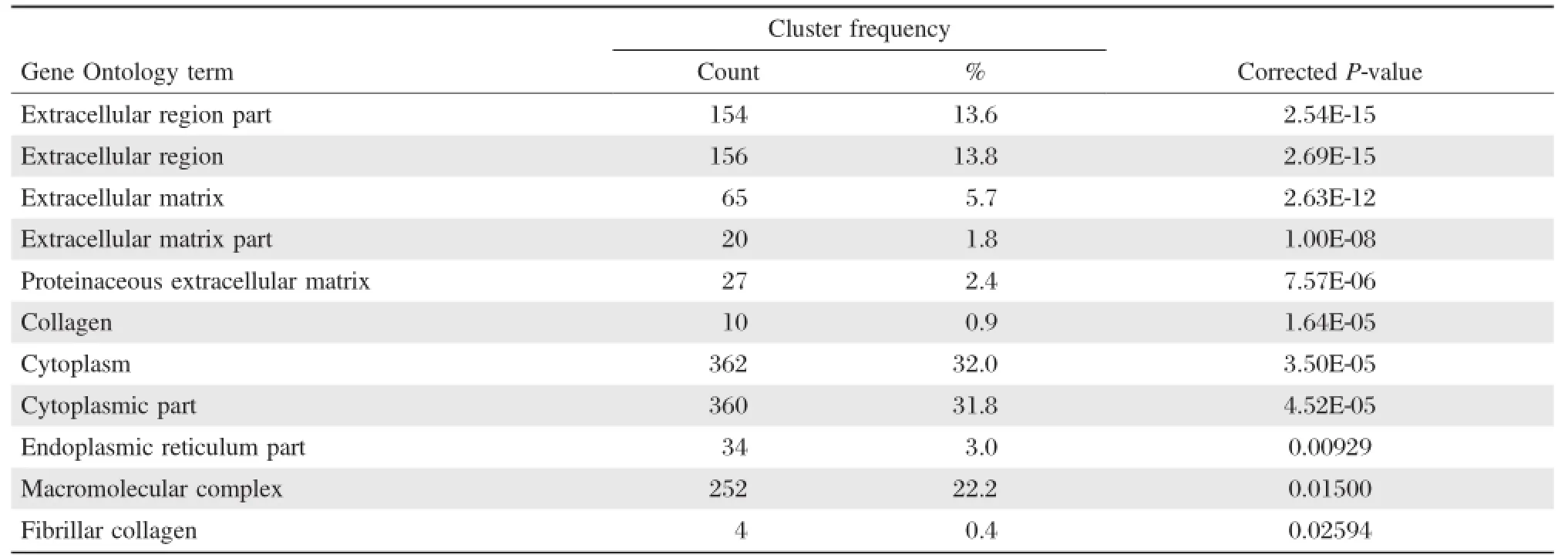
Table 3 Cellular distribution of the DEGs
GO functional enrichment analysis:biological processes
GO biological process analysis showed that the differentially expressed mRNAs were significantlyenriched in a total of 20 biological processes,including:extracellular structure organization,immune system process,and response to wounding(Table 5). The processes found to be most involved were the immune response,anatomical structure development, response to organic substance,developmental process, and response to stimulus.
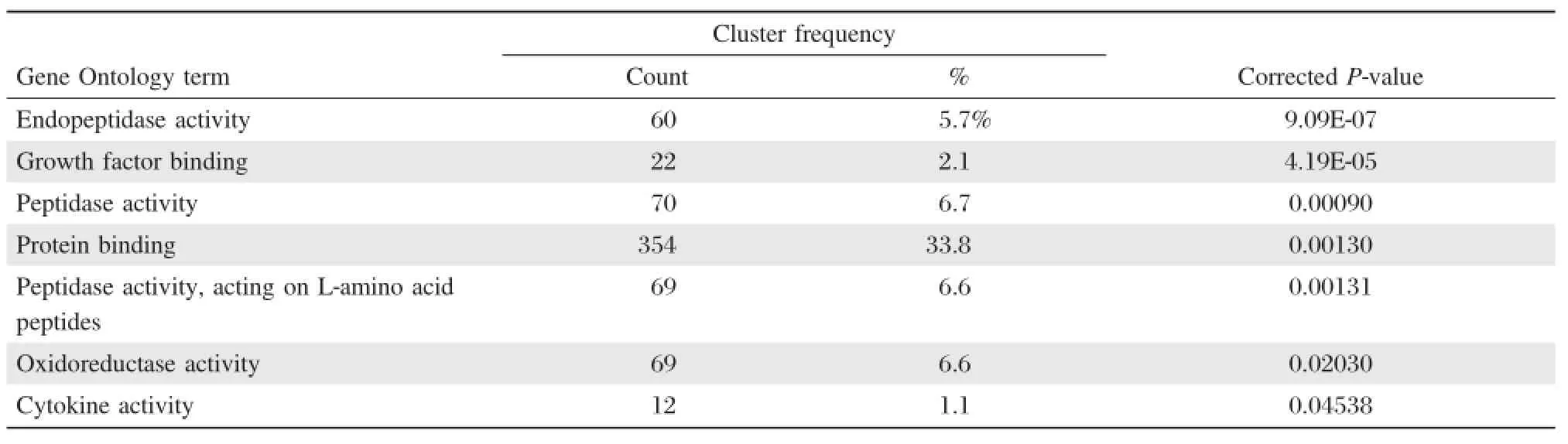
Table 4 Molecular function of the DEGs
Pathway enrichment analysis
The differentially expressed genes in the OSCC samples and matched paraneoplastic normal samples were involved in a total of 215 signaling pathways.Of these,68 signaling pathways were significantly enriched(Q-value≤0.05),e.g.,extracellular matrix(ECM)-receptor interaction,rheumatoid arthritis,and focal adhesion.Table 6shows the 10 most significantly enriched pathways.
Discussion
By utilizing high-throughput RNA-Seq technology, we have constructed genome-wide mRNA expression profiles in OSCC and detected 1,120 up-regulated mRNAs and 178 down-regulated mRNAs in OSCC tissue,compared with matched paraneoplastic normal tissue.These findings suggest that these aberrantly expressed mRNAs may play roles in OSCC development and progression.The most significantly up-regulated mRNAs was MAGEA11,which has been reported to be oncogenic.Bai et al.showed that high expression of MAGEA 11 increased the androgen receptor(AR)-dependent cell proliferation and promoted the development of prostate cancer[23].Lian et al.reported that MAGEA11 expression was more frequent in breast cancer tissue,compared to tumor-free breast tissue,suggesting that MAGEA11 may be involved in estrogen receptor (ER)-dependent cell proliferation and could act as a potential prognostic factor of poor outcome in breast cancer patients[24].Tsaia et al.reported differential expression of MAGEA11 in non-small cell lung cancer (NSCLC)[25].In contrast,the role of THCC,which was the most significantly down-regulated mRNA detected in our study,is unclear.In common with previous reports on mRNAs in OSCC,similar results were obtained in our study:MMP1(log2Ratio=1.038711)and MMP3 (log2Ratio=3.488456)have been reported as highly expressed in OSCC,and put forward as robust diagnostic biomarkers[26].PLAU(log2Ratio=1.925736)has been implicated in enhanced cell proliferation and migration and,has been proposed as a prognostic marker of relapse-free survival of OSCC,together with its receptor uPAR[27].Sapkota et al.reported that S100A14(log2Ratio= -1.595579)was progressively down-regulated in vitro during the transition from normal to dysplastic stages of cancer in OSCC progression.In addition,an inverse correlation between mRNA expression levels of MMP1 and MMP9 with S100A14 was found in 19 cases of OSCC[28].Other differentially expressed mRNAs identified in our study have been also been reported in different cancers,including AZGP1(log2Ratio= 8.392317),COL1A 1(log2Ratio=2.460066),SPARC (log2Ratio=1.866150),COL4A 1(log2Ratio= 1.487532),ISG15(log2Ratio=1.434671),RUNX 3(log2Ratio=1.430360),and KRT19(log2Ratio= -2.864896)[29-31].
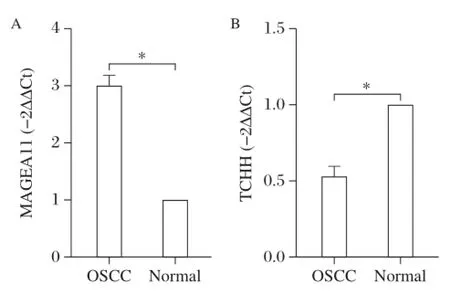
Fig.1 Expression of MAGEA11(A)and TCHH(B)in OSCC tissues.A:MRNA transcript levels MAGEA11 and B:TCHH in oral squamous cell carcinoma(OSCC)tissues n=15,*P<0.05 vs.normal controls.
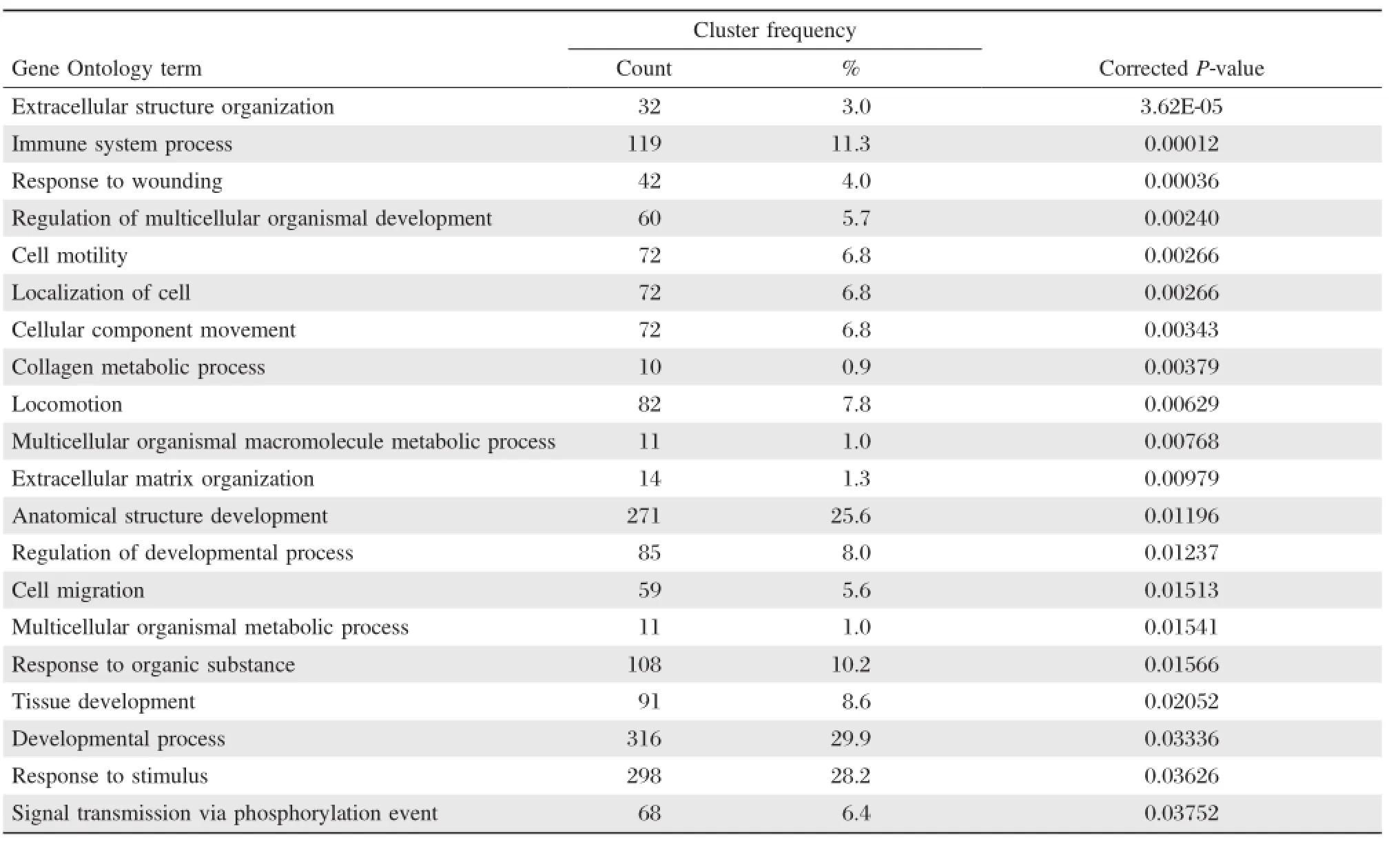
Table 5 Biological process associated with DEGs
Gene Ontology(GO)is an international standardized gene functional classification system which offers a dynamic-updated vocabulary to comprehensively describe properties of genes and their products in any organism.GO has three ontologies:molecular function, cellular component,and biological process.Every GO term belongs to one of these ontologies[32].Because genes often cooperate with each other to perform their biological functions,pathway analyses facilitate further understanding genes and their roles[33].
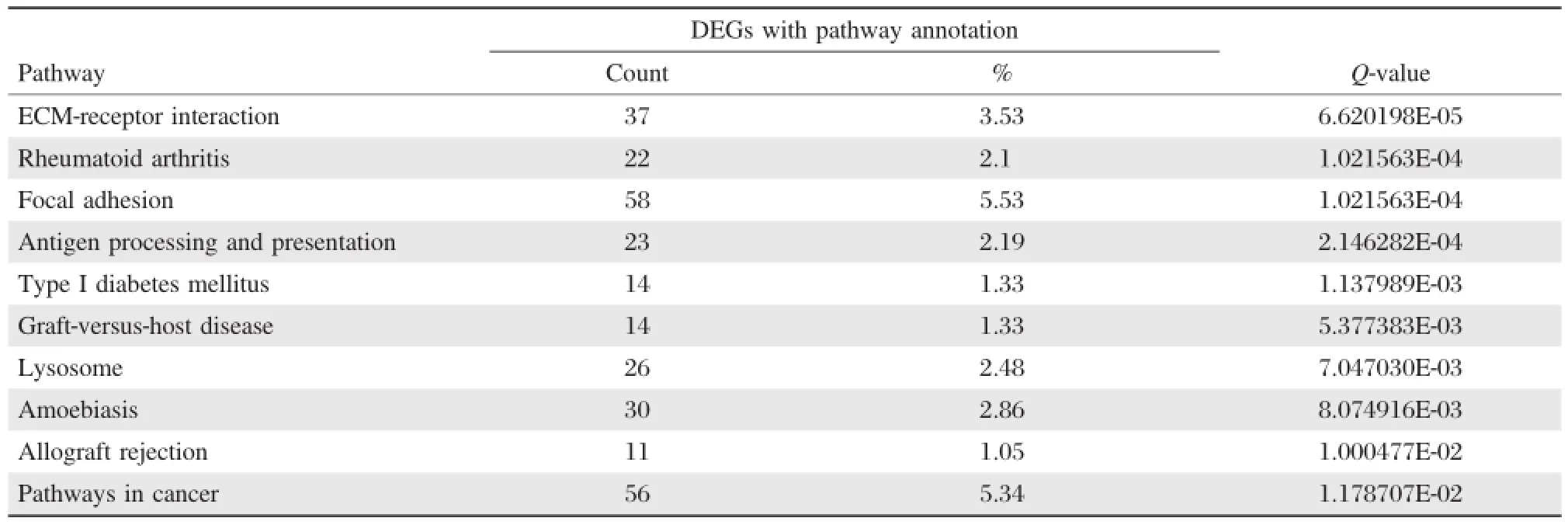
Table 6 The DEGs with pathway annotation
In this study,the results of GO functional enrichment analyses indicated that differentially expressed mRNAs in OSCC:were significantly enriched in cellular components,including extracellular region,extracellular matrix,macromolecular complex and cytoplasm; involved molecular functions that included endopeptidase activity,growth factor binding,peptidase activity; and may be involved in biological processes which areclosely related to the development and progression of cancer,including immune system process,cell migration,cell motility[34-35].These results suggest that these differentially expressed mRNAs play an important role in the tumorigenesis and development of OSCC.The results have provided an experimental basis for further research of these differentially expressed mRNAs in OSCC.
As shown inTable 6,the differentially expressed genes(DEGs)were enriched in 215 pathways, of which 68 were found to be significantly enriched by KEGG pathway enrichment analysis.These included ECM-receptor pathway interaction and focal adhesion pathways,which have both been previously reported in OSCC[29-31].In addition,the results of our study support previous studies which reported that mTOR affects the progression of OSCC,suggesting that the mTOR signaling pathway may play a role in OSCC development[36-37].
To our knowledge,our study presents the first genome-wide profiling of mRNAs of OSCC by high-throughput RNA-Seq.We utilized this data to identify the mRNAs that were significantly up-regulated or down-regulated in OSCC tissue compared to matched paraneoplastic normal tissue.By exploring their GO ontologies,our findings suggest that these differentially expressed mRNAs may act as oncogenes or tumor suppressors in the development and progression of OSCC.Our data has provided novel insights into cancer biology. However,further studies are required to determine their potential roles as diagnostic biomarkers of candidate therapeutic targets for OSCC.
Acknowledgments
This work was supported by a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions(PAPD,2014-37).
[1] Pisani P,Parkin DM,Ferlay J.Estimates of the worldwide mortality from eighteen major cancers in 1985.Implications for prevention and projections of future burden[J].Int J Cancer,1993,55(6):891-903.
[2] Benjamini Y,Yekutieli D.The control of the false discovery rate in multiple testing under dependency[J].Behav Brain Res,2001,29(4):1165-1188.
[3] Jemal A,Siegel R,Ward E,et al.Cancer statistics, 2009[J].CA Cancer J Clin,2009,59(4):225-249.
[4] Kang MK,Park NH.Conversion of normal to malignant phenotype:telomere shortening,telomerase activation,and genomic instability during immortalization of human oral keratinocytes[J].Crit Rev Oral Biol Med,2001,12(1):38-54.
[5] Williams HK.Molecular pathogenesis of oral squamous carcinoma[J].Mol Pathol,2000,53(4):165-172.
[6] Chu F,Feng Q,Qian Y,et al.ERBB2 gene amplification in oral squamous cell malignancies:a correlation with tumor progression and gene expression[J].Oral Surg Oral Med Oral Pathol Oral Radiol Endod,2011,112(1):90-95.
[7] Pradhan S,Nagashri MN,Gopinath KS,et al.Expression profiling of CYP1B1 in oral squamous cell carcinoma: counterintuitive downregulation in tumors[J].PLoS One, 2011,6(11):e27914.
[8] Gomes CC,Bernardes VF,Diniz MG,et al.Anti-apoptotic gene transcription signature of salivary gland neoplasms[J]. BMC Cancer,2012,12:61.
[9] Yasumatsu R,Nakashima T,Wakasaki T,et al.Relative level of thymidy late synthase mRNA expression in primary tumors and normal tissues predicts survival of patients with oral tongue squamous cell carcinoma[J]. Eur Arch Otorhinolaryngol,2010,267(4):581-586.
[10]Lodi G,Franchini R,Bez C,et al.Detection of survivin mRNA in healthy oral mucosa,oral leucoplakia and oral cancer[J].Oral Dis,2010,16(1):61-67.
[11]Zimmermann BG,Wong DT.Salivary mRNA targets for cancer diagnostics[J].Oral Oncol,2008,44(5):425-429.
[12]International Human Genome Consortium.Finishing the euchromatic sequence of the human genome[J].Nature, 2004,431(7011):931-945.
[13]Sultan M,Schulz MH,Richard H,et al.A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome[J].Science,2008,321(5891): 956-960.
[14]Metzker M L.Sequencing technologies-the next generation[J].Nat Rev Genet,2010,11(1):31-46.
[15]Xu W,Seok J,Mindrinos MN,et al.Human transcriptome array for high-throughput clinical studies[J].Proc Natl Acad Sci U S A,2011,108(9):3707-3712.
[16]Li X,Chen J,Hu X,et al.Comparative mRNA and microRNA expression profiling of three genitourinary cancers reveals common hallmarks and cancer-specific molecular events[J].PLoS One,2011,6(7):e22570.
[17]Zhu J,Jiang Z,Gao F,et al.A systematic analysis on DNA methylation and the expression of both mRNA and microRNA in bladder cancer[J].PLoS One,2011, 6(11):e28223.
[18]Morrissy AS,Morin RD,Delaney A,et al.Next-generation tag sequencing for cancer gene expression profiling[J]. Genome Res,2009,19(10):1825-1835.
[19]Hoen PA,Ariyurek Y,Thygesen HH,et al.Deep sequencing-based expression analysis shows major advances in robustness,resolution and inter-lab portability over five microarray platforms[J].Nucleic Acids Res, 2008,36(21):e141.
[20]Audic S,Claverie JM.The significance of digital gene expression profiles[J].Genome Res,1997,7(10):986-995.
[21]Benjamini Y,Yekutieli D.The control of the false discovery rate in multiple testing under dependency[J].Behav Brain Res,2001,29(4):1165-1188.
[22]Kanehisa M,Araki M,Goto S,et al.KEGG for linking genomes to life and the environment[J].Nucleic Acids Res,2008,36(Database issue):D480-D484.
[23]Bai S,Wilson EM.Epidermal-growth-factor-dependent phosphorylation and ubiquitinylation of MAGE-11 regulates its interaction with the androgen receptor[J].Mol Cell Biol,2008,28(6):1947-1963.
[24]Lian Y,Sang M,Ding C,et al.Expressions of MAGEA10 and MAGE-A11 in breast cancers and their prognostic significance:a retrospective clinical study[J].J Cancer Res Clin Oncol,2012,138(3):519-527.
[25]Tsai JR,Chong IW,Chen YH,et al.Differential expression profile of MAGE family in non-small-cell lung cancer[J].Lung Cancer,2007,56(2):185-192.
[26]Stott-Miller M,Houck JR,Lohavanichbutr P,et al.Tumor and salivary matrix metalloproteinase levels are strong diagnostic markers of oral squamous cell carcinoma[J].Cancer Epidemiol Biomarkers Prev,2011,20(12):2628-2636.
[27]Hundsdorfer B,Zeilhofer HF,Bock KP,et al.Tumourassociated urokinase-type plasminogen activator(uPA) and its inhibitor PAI-1 in normal and neoplastic tissues of patients with squamous cell cancer of the oral cavity-clinical relevance and prognostic value[J].J Cranio-maxillo fac Surg,2005,33(3):191-196.
[28]Sapkota D,Bruland O,Costea DE,et al.S100A14 regulates the invasive potential of oral squamous cell carcinoma derived cell-lines in vitro by modulating expression of matrix metalloproteinases,MMP1 and MMP9[J].Eur J Cancer,2011,47(4):600-610.
[29]Suhr ML,Dysvik B,Bruland O,et al.Gene expression profile of oral squamous cell carcinomas from Sri Lankan betel quid users[J].Oncol Rep,2007,18(5):1061-1075.
[30]Supic G,Kozomara R,Jovic N,et al.Hypermethylation of RUNX3 but not WIF1 gene and its association with stage and nodal status of tongue cancers[J].Oral Dis, 2011,17(8):794-800.
[31]Vincent-Chong V,Ismail S,Rahman Z,et al.Genome-wide analysis of oral squamous cell carcinomas revealed over expression of ISG15,Nestin and WNT11[J].Oral Dis,2012,18(5):469-476.
[32]Ashburner M,Ball CA,Blake JA,et al.Gene Ontology: tool for the unification of biology.The Gene Ontology Consortium[J].Nat Genet,2000,25(1):25-29.
[33]Kanehisa M,Goto S,Furumichi M,et al.KEGG for representation and analysis of molecular networks involving diseases and drugs[J].Nucleic Acids Res,2010, 38(Database issue):355-360.
[34]Menon R,Omenn GS.Proteomic characterization of novel alternative splice variant proteins in human epidermal growth factor receptor 2/neu-induced breast cancers[J]. Cancer Res,2010,70(9):3440-3449.
[35]Zeeberg BR,Reinhold W,Snajder R,et al.Functional categories associated with clusters of genes that are co-expressed across the NCI-60 cancer cell lines[J].PLoS One,2012,7(1):e30317.
[36]Naruse T,Kawasaki G,Yanamoto S,et al.Immunohistochemical study of VEGF expression in oral squamous cell carcinomas:correlation with the mTORHIF-1alpha pathway[J].Anticancer Res,2011,31(12):-4429-4437.
[37]Vitale-Cross L,Molinolo AA,Martin D,et al.Metformin prevents the development of oral squamous cell carcinomas from carcinogen-induced premalignant lesions[J]. Cancer Prev Res(Phila),2012,5(4):562-573.
△These authors contributed equally to this work.
✉Corresponding author:Pr of .Hongwei Li,Jiangsu Key Laboratory of Oral Diseases,Nanjing Medical University,Department of Oral and Maxillofacial Surgery,Affiliated Hospital of Stomatology,Nanjing Medical University, 136 Hanzhong Road,Nanjing,Jiangsu 210029,China,Tel:+86-25-
85031921;Fax:+86-25-85031921,Email address:lhwqhxa@sina.com. Received 22 June 2014,Revised 12 August2014,Accepted 22 November 2014,Epub 30 January 2015
R739.8,Document code:A
The authors reported no conflict of interests.
©2015 by the Journal of Biomedical Research.All rights reserved.
10.7555/JBR.29.20140088
猜你喜欢
趣味(语文)(2021年9期)2022-01-18 05:52:42
数学小灵通·3-4年级(2020年9期)2020-10-27 03:26:16
中成药(2018年9期)2018-10-09 07:18:42
中成药(2018年7期)2018-08-04 06:04:24
中成药(2018年7期)2018-08-04 06:04:06
红土地(2016年3期)2017-01-15 13:45:22
中国卫生(2016年10期)2016-11-13 01:07:44
幼儿智力世界(2016年6期)2016-05-14 13:50:51
发明与创新(2016年33期)2016-04-16 16:32:25
中国卫生(2015年10期)2015-11-10 03:14:32
 THE JOURNAL OF BIOMEDICAL RESEARCH2015年5期
THE JOURNAL OF BIOMEDICAL RESEARCH2015年5期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Serum IL-1βand IL-18 correlate with ESR and CRP in m ultid rug-resistant tuberculosis patients
- A simple practical balloon anchoring technique within the guide catheter for chronic total occlusion(CTO) of the coronary artery
- Prostate-specific antigen doubling time and response to cabazitaxel in a hormone-resistant metastatic prostate cancer patient
- Assessment of phytochemicals and antioxidant activities of raw and germinating Ceiba pentandra(kapok)seeds
- Diacerein protects against iodoacetate-induced osteoarthritis in the femorotibial joints of rats
- Acute effect of aspartame-induced oxidative stress in Wistar albino rat brain