
单分子实时测序技术的原理与应用
2015-02-04 06:26:32柳延虎王璐于黎
遗传 2015年3期
柳延虎,王璐,2,于黎,2
单分子实时测序技术的原理与应用
柳延虎1,王璐1,2,于黎1,2
1. 云南大学,云南省生物资源保护与利用重点实验室,昆明 650091;2. 云南大学,云南省高校动物遗传多样性与进化重点实验室,昆明 650091
单分子DNA测序技术是近10年发展起来的新一代测序技术,也称为第三代测序技术,包括单分子实时测序、真正单分子测序、单分子纳米孔测序等技术。文章介绍了单分子实时(Single-molecule real-time,SMRT)测序技术的基本原理、性能以及应用。与Sanger测序法和下一代测序技术相比,SMRT测序具有超长读长、测序周期短、无需模板扩增和直接检测表观修饰位点等特点,为研究人员提供了新选择。同时,SMRT测序的低准确率备受争议(约85%),其中约93%的错误是插入缺失,因此,其数据应用于基因组组装前需先对数据进行纠错处理。目前,SMRT测序在小型基因组从头测序和完整组装中已有良好应用,并且已经或将在表观遗传学、转录组学、大型基因组组装等领域发挥其优势,促进基因组学的研究。
单分子测序;PacBio;SMRT测序
DNA序列蕴藏了生物绝大部分遗传信息,是生物遗传和进化的基础。获得DNA序列对于阐明生命奥秘至关重要。为了测定DNA序列,1977年,Maxam和Gibert发明了化学降解法[1]。同年,Sanger发明了双脱氧末端终止法[2],即至今广泛应用的Sanger测序法。20世纪90年代,荧光自动测序技术用荧光代替Sanger法中的同位素,实现了自动化测序。这些技术现在也被称为第一代测序技术(First-generation sequencing)。应用Sanger测序法,人们完成了人类基因组计划。目前应用最广泛的第一代测序仪是ABI 3730xl测序仪,该测序仪拥有较长读长(平均读长700 bp)和极高准确率(99.9%),但是由于相对高昂的成本(表1),目前主要应用于细菌基因组测序、质粒测序、细菌人工染色体末端测序、突变位点验证等研究,而在大型基因组组装方面已很少应用。
近10年,下一代测序(Next-generation sequencing,NGS)技术相继出现并发展成熟,主要包括Roche公司的焦磷酸测序技术(454)[7]、Illumina公司的Solexa测序技术[8]和ABI公司的SOLiD测序技术[9],这些也被称为第二代测序技术(Second-generation sequencing)。第二代测序技术以高通量、低成本为主要特点(表1)。其中Hiseq 2500测序仪的通量达1 Tb,测序成本为30美元/Gb,与3730xl测序仪相比,单碱基成本降低到万分之一。第二代测序技术极大地推进了基因组学的发展,更多物种的基因组组装、重测序、甲基化、转录本、宏基因组等研究得以展开[10]。2012年11月,大型国际科研合作项目“千人基因组计划”的研究人员发布了1 092个人的基因组数据[11],该研究应用第二代测序技术完成了对世界上主要人群的基因组测序工作,绘制了迄今为止最详尽、最有医学应用价值的人类基因组遗传多态性图谱。另外,万种脊椎动物基因组计划[12]和万种微生物基因组计划(http://www.genomics.cn/news/show_news?nid=93218)正在进行中,这为生命科学和全球动物保护提供前所未有的基础资源。然而,读长相对较短仍是第二代测序技术的主要瓶颈。Roche公司454 GS FLX+测序仪平均测序读长最长,也仅为700 bp。Illumina公司Hiseq 2500测序仪的读长只有2×125 bp(表1)。较短的测序读长为组装基因组带来巨大困难;同时,由于采用了模板扩增步骤,其在组装高GC含量基因组时尤其受限[13]。
为了更好地发掘DNA序列信息,研究人员研发出最新一代测序方法——单分子的测序技术(Single- molecule sequencing),也称为第三代测序技术(Third- generation sequencing),其共同特征是基于单分子水平的边合成边测序,主要包括Pacific Biosciences (PacBio)公司的单分子实时(Single-molecule real- time,SMRT)测序技术、Oxford Nanopore公司的单分子纳米孔测序技术(The single-molecule nanopore DNA sequencing)、和Helicos公司的真正单分子测序技术(True single-molecule sequencing,tSMSTM)等[14]。目前,大部分第三代测序技术尚处于研发阶段,只有PacBio公司的RS(Real-timesequencing)系列测序仪已经商业化,其采用的就是SMRT测序技术。SMRT测序技术具有超长读长,还拥有不需要模板扩增、运行时间较短、直接检测表观修饰位点、较高的随机测序错误等特点。它弥补了第二代测序读长短、受GC含量影响大等局限性,已在小型基因组从头测序和组装中有较多应用。本文主要介绍了PacBio RS系列测序仪使用的SMRT测序技术的原理、性能及其应用。

表1 常用测序仪性能比较
注:#以测定96个样品为一次运行,其准确率为经过人工矫正后的准确率;##使用SBS v4试剂盒并且同时测两个flow cell为一次运行;*以测定8个SMRT cell为一次运行;**http://www.appliedbiosystems.com/absite/us/en/home/applications-technologies/solid- next-generation- sequencing/next-generation-systems/5500xl-solid.html。
1 SMRT测序原理
SMRT测序即单分子实时测序,其原理是:当DNA模板被聚合酶捕获后,4种不同荧光标记的dNTP通过布朗运动随机进入检测区域并与聚合酶结合,与模板匹配的碱基生成化学键的时间远远长于其他碱基停留的时间。因此统计荧光信号存在时间的长短,可区分匹配的碱基与游离碱基。通过统计4种荧光信号与时间的关系图(http://www.pacificbiosciences.com/),即可测定DNA模板序列。
SMRT测序核心技术之一是零级波导技术(Zero mode waveguide,ZMW)[15]。ZMW是一个直径只有10~50 nm的孔,远小于检测激光的波长(数百纳米)。因此当激光打在ZMW底部时,激光无法穿过,而是在ZMW底部发生衍射,只能照亮很小的区域。DNA聚合酶就被固定在这个区域。只有在这个区域内,碱基携带的荧光基团才能被激活而被检测到,大幅地降低了背景荧光干扰。每个ZMW只固定一个DNA聚合酶,当一个ZMW结合少于或超过一个DNA模板时,该ZMW所产生的测序结果在后续数据分析时被过滤掉,由此保证每个可用的ZMW都是一个单独的DNA合成体系。15万个ZMW聚合在一个芯片上,称为一个SMRT Cell。PacBio RSⅡ测序仪一个流程内可同时完成8个SMRT Cell的测序,产生3.2 Gb的数据(表1)。
SMRT测序的另一个核心技术是荧光基团标记在核苷酸3¢端磷酸上[16]。在DNA合成过程中,3¢端的磷酸键随着DNA链的延伸被断开,标记物被弃去,减少了DNA合成的空间位阻,维持DNA链连续合成,延长了测序读长。而第二代测序技术中荧光基团都标记在5¢端甲基上,在合成过程中,荧光标记物保留在DNA链上,随DNA链的延伸会产生三维空间阻力,导致DNA链延长到一定程度后出现错读,这是限制二代测序读长的原因之一。SMRT测序最大限度地保持了聚合酶的活性,是最接近天然状态的聚合酶反应体系。在实时监控系统下,DNA链以每秒10个碱基的速度合成。从建库到测序,整个过程在2 d内完成。
2 SMRT测序特点
2.1 超长读长
2013年4月,PacBio公司推出了PacBio RSⅡ测序仪,平均读长达到4 600 bp,最长读长超过 20 000 bp,每个SMRT cell 的通量为400 Mb。PacBio RSⅡ超长的读长非常有利于基因组组装,还可以填补已知基因组上的未测通区域,同时还开创了全新的应用领域:转录本全长测序和全长16S基因测序等[17, 18]。转录本全长测序为基因可变剪接形式的识别、复杂的转录分析和新基因探索提供了更有效的支持[19]。如Treutlein等[20]使用SMRT测序技术发现轴突蛋白基因家族中数百种不同亚型,这些基因产物展现出了惊人的复杂性,并提供了轴突蛋白在促进大脑细胞连接过程中作为识别分子具有重要作用的证据。
2.2 无需模板扩增
基因组GC含量直接影响DNA序列的测定。第二代测序技术中文库构建和测序过程中都有PCR 扩增步骤,高GC或低GC含量的基因组区域不容易被PCR扩增,导致在测序过程中测序覆盖度不足[21]。另外,在文库构建时需将DNA 打断成适当大小的片段,由于高GC含量区域不易被打断,使得这些片段过大而在长度筛选时被舍弃[21~23]。由于SMRT测序是真正意义上的单分子测序技术,没有PCR扩增步骤[6, 24],结合SMRT测序超长读长的特点,可以完成长片段的高GC含量区域测序,从而帮助高GC含量基因组完成组装。SMRT测序这一优势很好的应用到极端微生物的基因组研究中[13]。同时,无需模板扩增步骤还避免了PCR引入的错误,并且只需要使用极少的荧光基团,为今后大幅降低测序试剂成本提供了空间。
2.3 较短的运行时间
与第二代测序技术相比,SMRT测序简化了建库和测序步骤。一张SMRT cell从文库制备到测序完成只需要不到1 d时间。SMRT测序的读取速度可达每秒钟10个碱基,实现了DNA聚合酶自身反应速度,大幅缩短了测序周期,因此可以在酶失活之前测得更长的序列。较短的运行时间对于应对传染病爆发尤为重要,在很短时间内得到变异微生物的基因组,可以为快速和准确地研究爆发起因以及治疗策略提供基础[25]。
2.4 直接检测表观修饰位点
SMRT测序在DNA合成时,如遇到模板上的甲基化碱基,则从dNTP与DNA聚合酶结合至释放荧光基团的时间显著大于遇到非甲基化碱基所需时间,并且不同类型的修饰碱基具有不同的DNA聚合酶动力学特征。最终根据这些动力学特征,主要是脉冲间隔时间(Interpulse duration,IPD)长短,判断碱基的甲基化类型[16]。因此,在较高测序覆盖度(>15×)的前提下,SMRT测序可在完成常规测序的同时,还能获得5-甲基胞嘧啶、5-羟甲基胞嘧啶和N6-甲基腺嘌呤的信息[26]。因为5-甲基胞嘧啶可能与5-羟甲基胞嘧啶执行不同的生物功能[27],所以如何简单且有效区分这两种不同修饰碱基一直是分子生物学家关注的问题。应用SMRT测序技术,可以较好地解决这个问题。
2.5 直接测转录本
如果将DNA聚合酶换成RNA反转录酶,SMRT测序就可以直接进行RNA测序,无需逆转录[28]。这一技术目前尚处于研发阶段。RNA直接测序将降低体外逆转录产生的系统误差,还可以检测RNA上碱基的分子修饰,这将打开一个全新的研究思路。Uemura等[29]使用该技术对核糖体中mRNA的翻译过程进行了实时测序观察,实时观测到了单个核糖体如何将氨基酸串联起来的过程。
2.6 较高但是完全随机的测序错误
SMRT测序的错误率大约是15%,碱基错测率约1%,其他错误主要是单碱基的插入和缺失(Insertions and deletions,INDELs)。但是当覆盖度超过15×时,SMRT测序过程中产生的错误通过概率算法进行纠正后,其正确率可达99.3%[16]。通过纠错提高序列准确性需要较高覆盖度和大量计算机资源,这对大多数动植物大型基因组来说较难实施,最根本的解决方法是通过技术革新提升SMRT测序本身的准确率。值得注意的是,SMRT测序的错误都是随机错误,而非系统错误,系统错误是无法通过提高测序覆盖度矫正的[16]。
3 SMRT测序数据分析软件
为了更好地应用SMRT测序技术所产生的序列数据,近几年报道了一些应用这些数据的算法及软件(表2)。从表2可以看出,这些软件涵盖了组装软件(HGAP[30]、ALLPATHS-LG[31]、AHA[32]、MIRA[33]、PacBioToCA[34])、比对软件(BLASR[35])、纠错软件(LSC[36]、PacBioToCA)、补“洞”软件(AHA、PBJelly[37])、数据模拟软件(PBSIM[38])(表2)。
由于SMRT测序数据高错误率的特点,应用这些数据组装基因组和补“洞”前需要先纠正其中的错误。根据纠错过程是否使用第二代测序数据可将软件分成两类。目前,利用第二代测序数据的高准确度数据纠正SMRT数据的软件有LSC、PacBioToCA等,这两种方法虽然可以将SMRT测序数据的错误率降到小于1%,但是纠错的同时将第二代测序数据的系统错误引入其中,并且需要消耗大量的计算机资源。为了避免使用第二代测序数据,Chin等[30]开发了软件HGAP,该软件使用SMRT测序数据中的较短序列纠正较长的“种子”序列,再用纠错后的“种子”长序列组装基因组。HGAP软件使用SMRT测序数据独立地完成纠错和组装,不再依赖第二代测序数据。目前,这个软件适用于组装小于130 M的基因组。在这些组装软件中,软件AHA更适合搭建基因组草图框架,其余几个组装软件更适合混合拼接。BLASR软件是快速比对软件,适用于将SMRT测序数据比对到基因组,因为BLASR比其他比对软件更好地“容忍”这些数据中的INDEL。由于基因组上存在重复区域和高GC含量区域,目前已公布的基因组都存在“洞”,SMRT测序超长读长和无GC偏好可以很好地完善基因组,可以应用PBJelly等软件填补基因组上的“洞”。

表2 SMRT数据的分析软件
4 SMRT测序应用
目前SMRT测序技术已应用在基因组组装、转录组测序、甲基化分析和基因组重测序等方面(表3),尤其是在基因组组装和甲基化研究中有着独特优势,下面将详细介绍这两个方面的应用。
4.1 组装基因组
4.1.1 大型基因组组装
SMRT测序具有超长的读长,对于组装大型基因组很有帮助。但是,由于其价格较高,通常用第二代测序数据加SMRT测序数据混合组装的策略组装大型基因组。Koren等[34]对比了第二代测序数据和混合数据组装的虎皮鹦鹉()基因组结果:用Illumina 数据194×(包括短片段文库和长片段文库)组装的conitg 数目为24 181,N50为47 838 bp;用454数据15.4×(包括短片段文库和长片段文库) 组装得到contig 16 574条,N50长度为75 178 bp;用454数据15.4×加上PacBioToCA纠错后的SMRT测序数据3.83×混合组装,conitg数目为15 328条,N50 长度为93 069 bp。PacBio数据和454数据混合组装与仅用454数据组装的结果相比,conitg数目减少了1 246条,N50提升了24%。由此可见,SMRT测序超长的读长能帮助提高大型基因组组装效果。
4.1.2 小型基因组组装
与混合组装大型基因组不同,单独使用SMRT测序数据即可很好地完成小型基因组组装。Chin等[30]比较了仅用SMRT测序数据组装和混合组装大肠杆菌基因组(4 639 675 bp):混合组装用130×Illumina数据和133×SMRT测序数据,采用ALLPATHS-LG软件组装得到1个contig,长度为4 638 970 bp;仅用99×SMRT测序数据,使用HGAP软件组装得到2个conitg,N50为4 648 564 bp,接近基因组全长。只用SMRT测序数据组装得到与混合组装相差无几的结果。应用SMRT测序数据组装小型基因组已有较多报道(表3)。
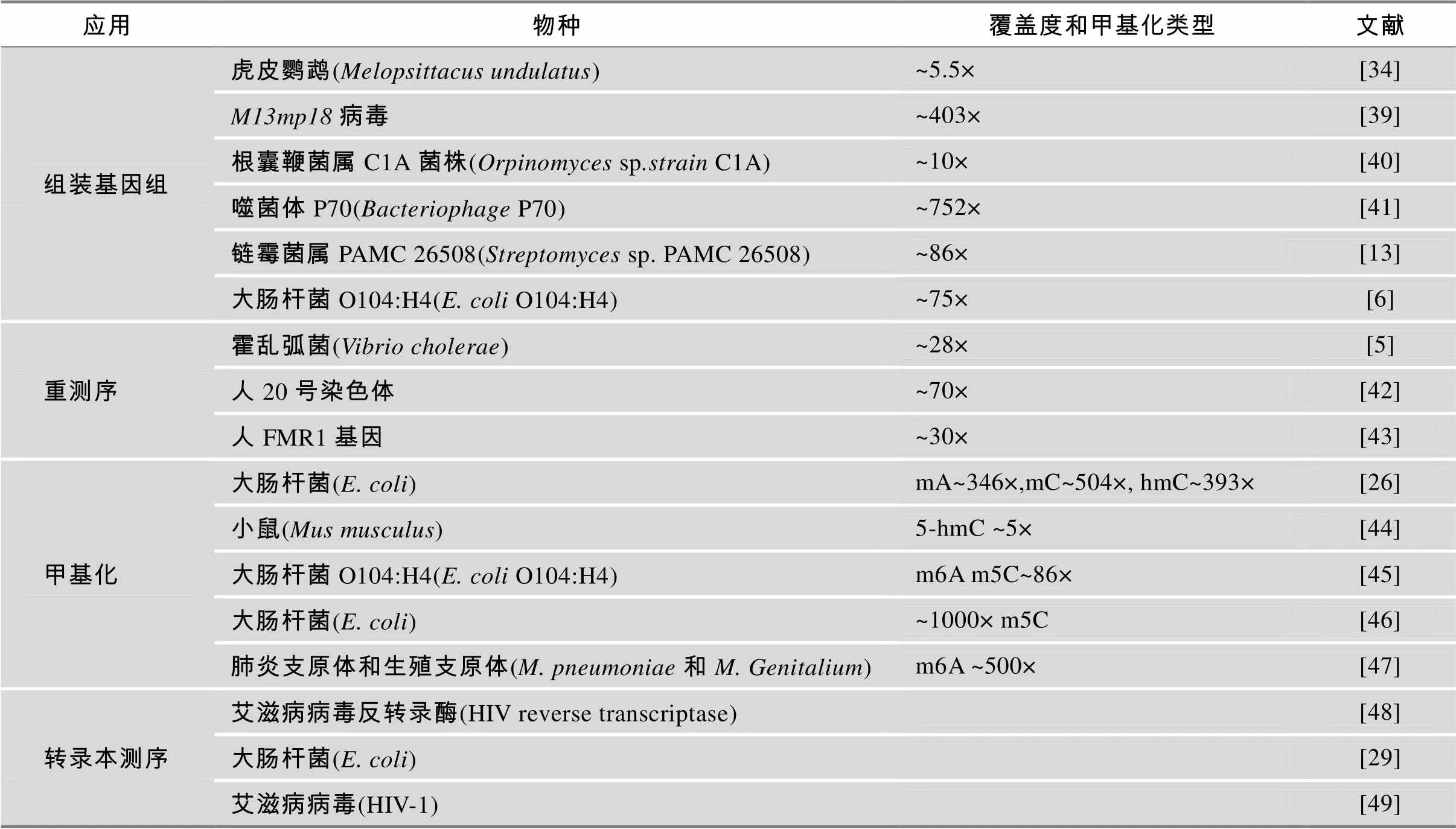
表3 SMRT测序在各方面的应用
PacBio公司在2013年1月加入100K基因组计划,该计划旨在测序100 000种食源性致病菌基因组,并在同年8月已完成20种食源性病原微生物的基因组测序工作。使用SMRT测序数据不但将每个基因组组装成了单个染色体,还包含了完整的表观遗传学信息、完整的噬菌体以及质粒元件信息。这对于了解菌株致病性、耐药性及其他与生存相关的重要生物性状非常关键。这些信息将有助于减少诊断和确定爆发株所需时间。目前已经将序列和完整的表观遗传学信息发布在NCBI网站(http://www. ncbi.nlm.nih.gov/bioproject/186441)。
4.1.3 高/低GC含量基因组组装
SMRT测序没有PCR扩增步骤,可以很好地完成高GC含量基因组的组装。韩国极地研究所Dr. Park研究团队首先利用Illumina Hiseq 2000平台对南极微生物sp. PAMC 26508的基因组进行测序[13]。该菌株基因组GC含量高达71%,即使利用Hiseq 2000平台进行了200×深度的测序,仍无法获得完整基因组。组装时产生了185 个contigs,随后使用Sanger法仍然无法有效填补草图中的缺口。研究人员利用14×纠错过的SMRT测序数据和8×454数据混合组装,得到5个染色体conitgs和1个质粒conitg,conitg N50达到1 430 884 bp。SMRT测序对高GC区域有着较其他测序仪更好的测序效果,是一项非常好的从头测序和组装高GC含量基因组的新工具。
SMRT测序也非常适用于组装低GC含量的叶绿体基因组[50]。Ferrarini等[51]使用9111×Illumina HiSeq2000数据组装的叶绿体基因组,只能组装到7个contigs,有9.41%的基因组未覆盖到。而使用320×的SMRT测序数据则将叶绿体组装成一个完整的基因组。文章中统计了SMRT测序所得序列与GC含量的相关性,结果显示SMRT测序无明显的GC偏好。
4.1.4 完善已有基因组
利用SMRT测序技术超长读长和无GC偏好的优势,可对模式生物基因组草图进行改善。Richard Gibbs团队开发了高度自动化的工具PBJelly,能够将SMRT测序所得长片段与基因组草图进行比对,填补或减少草图中的缺口,从而完善基因组草图[37]。他们用18×SMRT测序数据对黑腹果蝇基因组进行补洞,缺口数目从4 651个降低到311个,降低了15倍;conitg N50从64 006 bp提高到723 621 bp,提高了11倍。同时他们用6.8×SMRT测序数据对黑子白眉猴基因组(2.8 G)进行升级,缺口数目从186 841个降低到66 211个,降低了2.8倍;contig N50从34 925 bp提升到128 379 bp,提升了3.7倍。由此可见,应用SMRT测序提升基因组组装结果,不仅可以减少缺口数目,还可以大大提高contig的长度,而且对大型基因组和小型基因组提升效果同样明显。
4.2 甲基化分析
SMRT测序系统不需要进行重亚硫酸盐处理等额外实验步骤,就能够直接进行表观遗传学分析。New England Biolabs联合Pacific Biosciences的研究人员利用PacBio RS系统对6种细菌基因组进行了重测序[52],不仅鉴定出细菌基因组中新的胞嘧啶和腺嘌呤甲基化位点,还鉴定出介导这些表观遗传学标志的甲基转移酶。SMRT测序系统可以同时对碱基序列和碱基修饰两方面测序信息进行分析,这为表观遗传学及疾病基因组学开辟了新的研究思路。
5 结语与展望
近10年,DNA测序技术飞速发展,多种第二代和第三代测序仪相继问世。从第一代测序到第三代测序都有各自的优势。Sanger测序通量低、读长较长、准确率高,对于小量测序仍是最佳选择。高通量、低成本的第二代测序已发展成熟,在大型基因组测序和重测序中广泛应用。尤其是Illumina公司2014年推出的Hiseq X Ten测序仪,已实现1000美元完成一个人类基因组的目标(http://www.nature.com/news/is-the-1-000-genome-for-real-1.14530)。SMRT测序以超长的读长、无GC偏好等优势,可以完成高GC含量、重复区域多的基因组,已广泛应用到细菌和真菌基因组学研究中,并将在更多研究领域发挥作用。在未来的一段时间里,三代测序技术将共同存在,在不同的领域发挥各自优势,并互相补充[53]。鉴于第一代、第二代测序技术的一些局限性,人们越来越关注单分子测序。针对已商业化的SMRT测序技术,目前已公布了一系列相关的应用软件,涵盖了组装、纠错、比对、数据模拟和补洞等常用分析。SMRT测序在细菌和真菌的基因组学以及表观遗传学研究中已有较多应用,为解决生物学问题提供了新的方案。随着SMRT测序的不断发展,相信在不久的将来,它将为转录组学分析、大型基因组组装等领域提供有力的支持。
目前,未商用化的其他单分子测序技术有Helicos Biosciences公司的True single-molecule sequencing (tSMSTM)[54]、Oxford Nanopore Technologies公司的the single-molecule nanopore DNA sequencing (https: //nanoporetech.com/)、NABsys公司的‘Hybridization’-assisted nanopore sequencing (HANS)等 (http://www. nabsys.com/)。其中Oxford Nanopore Technologies公司将推出GridION和MinION两款基于纳米孔DNA测序技术的便携式基因组测序仪,后者仅有U盘大小,可插入计算机的USB端口完成测序工作,价格仅900美元。
2009年9月,中国科学院北京基因组研究所与浪潮集团共同成立了“中科院北京基因组所-浪潮基因组科学联合实验室”(http://www.big.ac.cn/ydhz/ hzxm/200909/t20090918_2511445.html),联合各领域科研力量共同研发单分子测序技术,目前尚处于研发阶段(http://www.big.ac.cn/ydhz/hzdt/200912/t20091207_ 2690938. html)。作为基因科学的战略性装备,具有国际先进水平的第三代DNA测序仪的研制,将为中国在该领域取得领先优势奠定基础。拥有自主知识产权的第三代测序仪不仅将填补中国在DNA测序基础装备领域的空白、提升装备自主化水平,同时也将使国内生命科学研究机构能获得低成本、高效率的测序工具。
随着单分子测序技术的不断发展及完善,预计单分子测序技术成本将逐渐下降。这将有利于展开个人基因组测序工作,基因组水平指导下的遗传病诊治、个人医疗和保健等工作可以更高效的进行,人们进入人性化医疗时代。同时也便捷了各领域的研究人员获得研究领域的物种基因组,促进基因组学研究的发展。
[1] Maxam AM, Gilbert W. A new method for sequencing DNA., 1977, 74(2): 560–564.
[2] Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors., 1977, 74(12): 5463–5467.
[3] Liu L, Li YH, Li SL, Hu N, He YM, Pong R, Lin DN, Lu LH, Law M. Comparison of next-generation sequencing systems., 2012, 2012: Article ID 251364.
[4] Gilles A, Meglécz E, Pech N, Ferreira S, Malausa T, Martin JF. Accuracy and quality assessment of 454 GS-FLX Titanium pyrosequencing., 2011, 12(1): 245.
[5] Chin CS, Sorenson J, Harris JB, Robins WP, Charles RC, Jean-Charles RR, Bullard J, Webster DR, Kasarskis A, Peluso P, Paxinos EE, Yamaichi Y, Calderwood SB, Mekalanos JJ, Schadt EE, Waldor MK. The origin of the Haitian cholera outbreak strain., 2011, 364(1): 33–42.
[6] Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D, Klammer A, Peluso P, Lee L, Kislyuk AO, Bullard J, Kasarskis A, Wang S, Eid J, Rank D, Redman JC, Steyert SR, Frimodt-Møller J, Struve C, Petersen AM, Krogfelt KA, Nataro JP, Schadt EE, Waldor MK. Origins of the E. coli strain causing an outbreak of hemolytic–uremic syndrome in Germany., 2011, 365(8): 709–717.
[7] Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen ZT, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu PG, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors., 2005, 437(7057): 376–380.
[8] Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu XH, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu XL, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara E Catenazzi M, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo SJ, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning ZM, Ling Ng B, Novo SM, O'Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang JW, Worsley GJ, Yan JY, Yau L, Zuerlein M, Rogers J, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ. Accurate whole human genome sequencing using reversible terminator chemistry., 2008, 456(7218): 53–59.
[9] Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, Sidow A, Fire A, Johnson SM. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning., 2008, 18(7): 1051–1063.
[10] 杨晓玲, 施苏华, 唐恬. 新一代测序技术的发展及应用前景. 生物技术通报, 2010, (10): 76–81.
[11] 1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1, 092 human genomes., 2012, 491(7422): 56–65.
[12] Haussler D, O'Brien S J, Ryder OA, Barker FK, Clamp M, Crawford AJ, Hanner R, Hanotte O, Johnson WE, McGuire JA, Miller W, Murphy RW, Murphy WJ, Sheldon FH, Sinervo B, Venkatesh B, Wiley EO, Allendorf FW, Amato G, Baker CS, Bauer A, Beja-Pereira A, Bermingham E, Bernardi G, Bonvicino CR, Brenner S, Burke T, Cracraft J, Diekhans M, Edwards S, Ericson PG, Estes J, Fjelsda J, Flesness N, Gamble T, Gaubert P, Graphodatsky AS, Marshall Graves JA, Green ED, Green RE, Hackett S, Hebert P, Helgen KM, Joseph L, Kessing B, Kingsley DM, Lewin HA, Luikart G, Martelli P, Moreira MA, Nguyen N, Ortí G, Pike BL, Rawson DM, Schuster SC, Seuánez HN, Shaffer HB, Springer MS, Stuart JM, Sumner J, Teeling E, Vrijenhoek RC, Ward RD, Warren WC, Wayne R, Williams TM, Wolfe ND, Zhang YP. Genome 10K: a proposal to obtain whole-genome sequence for 10 000 vertebrate species., 2009, 100(6): 659–674.
[13] Shin SC, Ahn do H, Kim SJ, Lee H, Oh TJ, Lee JE, Park H. Advantages of single-molecule real-time sequencing in high-GC content genomes., 2013, 8(7): e68824.
[14] 李明爽, 赵敏. 第三代测序基本原理. 现代生物医学进展, 2012, 12(10): 1980–1982.
[15] Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, Webb WW. Zero-mode waveguides for single-molecule analysis at high concentrations., 2003, 299(5607): 682–686.
[16] Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong XX, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma CC, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. Real-time DNA sequencing from single polymerase molecules., 2009, 323(5910): 133–138.
[17] Heiner C, Baybayan P, Wang S, Guo Y, Ashby M, Wilson J, Travers K, Chin J, Underwood J. Greater than 10 kb read lengths routine when sequencing with Pacific Biosciences’ XL release., 2013, 24(S): S43.
[18] Mosher JJ, Bowman B, Bernberg EL, Shevchenko O, Kan JJ, Korlach J, Kaplan LA. Improved performance of the PacBio SMRT technology for 16S rDNA sequencing., 2014, 104: 59–60.
[19] Sharon D, Tilgner H, Grubert F, Snyder M. A single-molecule long-read survey of the human transcriptome., 2013, 31(11): 1009–1014.
[20] Treutlein B, Gokce O, Quake SR, Südhof TC. Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing., 2014, 111(13): E1291-E1299.
[21] Aird D, Ross MG, Chen WS, Danielsson M, Fennell T, Russ C, Jaffe DB, Nusbaum C, Gnirke A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries., 2011, 12(2): R18.
[22] Niu BF, Fu LM, Sun SL, Li WZ. Artificial and natural duplicates in pyrosequencing reads of metagenomic data., 2010, 11: 187.
[23] Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-short read data sets from high- throughput DNA sequencing., 2008, 36(16): e105.
[24] Schadt EE, Turner S, Kasarskis A. A window into third- generation sequencing., 2010, 19(R2): R227-R240.
[25] 刘岩, 吴秉铨. 第三代测序技术: 单分子即时测序. 中华病理学杂志, 2011, 40(10): 718–720.
[26] Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. Direct detection of DNA methylation during single-molecule, real-time sequencing., 2010, 7(6): 461–465.
[27] Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain., 2009, 324(5929): 929–930.
[28] 张得芳, 马秋月, 尹佟明, 夏涛. 第三代测序技术及其应用. 中国生物工程杂志, 2013, 33(5): 125–131.
[29] Uemura S, Aitken CE, Korlach J, Flusberg BA, Turner SW, Puglisi JD. Real-time tRNA transit on single translating ribosomes at codon resolution., 2010, 464(7291): 1012–1017.
[30] Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data., 2013, 10(6): 563–569.
[31] Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, Berlin AM, Aird D, Costello M, Daza R, Williams L, Nicol R, Gnirke A, N J Virol usbaum C, Lander ES, Jaffe DB. High-quality draft assemblies of mammalian genomes from massively parallel sequence data., 2011, 108(4): 1513–1518.
[32] Bashir A, Klammer AA, Robins WP, Chin CS, Webster D, Paxinos E, Hsu D, Ashby M, Wang S, Peluso P, Sebra R, Sorenson J, Bullard J, Yen J, Valdovino M, Mollova E, Luong K, Lin S, LaMay B, Joshi A, Rowe L, Frace M, Tarr CL, Turnsek M, Davis BM, Kasarskis A, Mekalanos JJ, Waldor MK, Schadt EE. A hybrid approach for the automated finishing of bacterial genomes., 2012, 30(7): 701–707.
[33] Chevreux B. MIRA: an automated genome and EST assembler[Ph. D. Thesis]. Duisburg: Heidelberg, 2005.
[34] Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, Phillippy AM. Hybrid error correction and de novo assembly of single-molecule sequencing reads., 2012, 30(7): 693–700.
[35] Chaisson MJ, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory., 2012, 13(1): 238.
[36] Au KF, Underwood JG, Lee L, Wong WH. Improving PacBio long read accuracy by short read alignment., 2012, 7(10): e46679.
[37] English AC, Richards S, Han Y, Wang M, Vee V, Qu JX, Qin X, Muzny DM, Reid JG, Worley KC, Gibbs RA. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology., 2012, 7(11): e47768.
[38] Ono Y, Asai K, Hamada M. PBSIM: PacBio reads simulator—toward accurate genome assembly., 2013, 29(1): 119–121.
[39] Coupland P, Chandra T, Quail M, Reik W, Swerdlow H. Direct sequencing of small genomes on the Pacific Biosciences RS without library preparation., 2012, 53(6): 365–372.
[40] Youssef NH, Couger MB, Struchtemeyer CG, Liggenstoffer AS, Prade RA, Najar FZ, Atiyeh HK, Wilkins MR, Elshahed MS. The genome of the anaerobic fungussp. strain C1A reveals the unique evolutionary history of a remarkable plant biomass degrader., 2013, 79(15): 4620–4634.
[41] Schmuki MM, Erne D, Loessner MJ, Klumpp J. Bacteriophage P70: Unique morphology and unrelatedness to other Listeria bacteriophages., 2012, 86(23): 13099–13102.
[42] Carneiro MO, Russ C, Ross MG, Gabriel SB, Nusbaum C, DePristo MA. Pacific biosciences sequencing technology for genotyping and variation discovery in human data., 2012, 13(1): 375.
[43] Loomis EW, Eid JS, Peluso P, Yin J, Hickey L, Rank D, McCalmon S, Hagerman RJ, Tassone F, Hagerman PJ. Sequencing the unsequenceable: Expanded CGG-repeat alleles of the fragile X gene., 2013, 23(1): 121–128.
[44] Song CX, Clark TA, Lu XY, Kislyuk A, Dai Q, Turner SW, He C, Korlach J. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine., 2012, 9(1): 75–77.
[45] Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, Feng ZX, Losic B, Mahajan MC, Jabado OJ, Deikus G, Clark TA, Luong K, Murray IA, Davis BM, Keren-Paz A, Chess A, Roberts RJ, Korlach J, Turner SW, Kumar V, Waldor MK, Schadt EE. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing., 2012, 30(12): 1232–1239.
[46] Schadt EE, Banerjee O, Fang G, Feng ZX, Wong WH, Zhang XG, Kislyuk A, Clark TA, Luong K, Keren-Paz A, Chess A, Kumar V, Chen-Plotkin A, Sondheimer N, Korlach J, Kasarskis A. Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases., 2013, 23(1): 129–141.
[47] Lluch-Senar M, Luong K, Lloréns-Rico V, Delgado J, Fang G, Spittle K, Clark TA, Schadt E, Turner SW, Korlach J, Serrano L. Comprehensive methylome characterization ofat single-base resolution., 2013, 9(1): e1003191.
[48] Vilfan ID, Tsai YC, Clark TA, Wegener J, Dai Q, Yi CQ, Pan T, Turner SW, Korlach J. Analysis of RNA base modification and structural rearrangement by single-molecule real-time detection of reverse transcription., 2013, 11(1): 8.
[49] Ocwieja KE, Sherrill-Mix S, Mukherjee R, Custers-Allen R, David P, Brown M, Wang S, Link DR, Olson J, Travers K, Schadt E, Bushman FD. Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing., 2012, 40(20): 10345–10355.
[50] Li QS, Li Y, Song JY, Xu HB, Xu J, Zhu YJ, Li XW, Gao HH, Dong LL, Qian J, Sun C, Chen SL. High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy., 2014, 204(4): 1041–1049.
[51] Ferrarini M, Moretto M, Ward JA, Šurbanovski N, Stevanović V, Giongo L, Viola R, Cavalieri D, Velasco R, Cestaro A, Sargent DJ. An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome., 2013, 14(1): 670.
[52] Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, Fomenkov A, Turner SW, Korlach J, Roberts RJ. The methylomes of six bacteria., 2012, 40(22): 11450–11462.
[53] 王兴春, 杨致荣, 王敏, 李玮, 李生才. 高通量测序技术及其应用. 中国生物工程杂志, 2012, 32(1): 109–114.
[54] Harris TD, Buzby PR, Babcock H, Beer E, Bowers J, Braslavsky I, Causey M, Colonell J, DiMeo J, Efcavitch JW, Giladi E, Gill J, Healy J, Jarosz M, Lapen D, Moulton K, Quake SR, Steinmann K, Thayer E, Tyurina A, Ward R, Weiss H, Xie Z. Single-molecule DNA sequencing of a viral genome., 2008, 320(5872): 106–109.
(责任编委: 胡松年)
The principle and application of the single-molecule real-time sequencing technology
Yanhu Liu1, Lu Wang1,2, Li Yu1,2
Last decade witnessed the explosive development of the third-generation sequencing strategy, including single-molecule real-time sequencing (SMRT), true single-molecule sequencing (tSMSTM) and the single-molecule nanopore DNA sequencing. In this review, we summarize the principle, performance and application of the SMRT sequencing technology. Compared with the traditional Sanger method and the next-generation sequencing (NGS) technologies, the SMRT approach has several advantages, including long read length, high speed, PCR-free and the capability of direct detection of epigenetic modifications. However, the disadvantage of its low accuracy, most of which resulted from insertions and deletions, is also notable. So, the raw sequence data need to be corrected before assembly. Up to now, the SMRT is a good fit for applications in thegenomic sequencing and the high-quality assemblies of small genomes. In the future, it is expected to play an important role in epigenetics, transcriptomic sequencing, and assemblies of large genomes.
single molecule sequencing; PacBio; SMRT sequencing
2014-09-25;
2014-12-01
国家自然科学基金重大研究计划项目(编号:91131904)和中国科学院昆明动物研究所遗传资源与进化国家重点实验室开放课题(编号:GREKF14-04)资助
柳延虎,博士研究生,研究方向:基因组学。E-mail: liuyanhu005@163.com
于黎,研究员,博士生导师,研究方向:动物遗传与进化。E-mail: yuli-1220@163.com王璐,助理研究员,研究方向:基因组学。E-mail: wanglu@ynu.edu.cn
致 谢: 感谢王国栋在文章写作和修改过程中的指导和宝贵意见,感谢陈岳工程师在文章写作过程中给予的帮助。
10.16288/j.yczz.14-323
2015-1-19 16:51:26
http://www.cnki.net/kcms/detail/11.1913.R.20150119.1651.002.html
猜你喜欢
新民周刊(2022年27期)2022-08-01 07:04:49
今日农业(2021年11期)2021-08-13 08:53:24
传染病信息(2021年6期)2021-02-12 01:52:58
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
化学工业与工程(2015年1期)2015-02-10 03:01:41
遗传(2014年3期)2014-02-28 20:58:49
