
高纯度(98.5% )夫沙那韦-d4的合成*
2015-02-02 07:53:43沈加林沈小明吕爱娟陈礼勤南京地质矿产研究所江苏南京006南京靖龙医药技术有限公司江苏南京800
合成化学 2015年7期
关键词:合成
时 磊,沈加林,沈小明,吕爱娟,陈礼勤(.南京地质矿产研究所,江苏南京 006; .南京靖龙医药技术有限公司,江苏南京 800)
高纯度(98.5% )夫沙那韦-d4的合成*
时磊1,沈加林1,沈小明1,吕爱娟1,陈礼勤2
(1.南京地质矿产研究所,江苏南京210016; 2.南京靖龙医药技术有限公司,江苏南京211800)
摘要:以硝基苯-d5为原料,经还原、保护、磺酰化、脱保护、氧化和亲核取代反应制得中间体——对硝基苯磺酰氯-d4(7)。以(2R,3S) -N-叔丁氧羰基-3-氨基-1,2-环氧-4-苯基丁烷为原料,经开环反应制得(1S,2R) -N-[1-苯甲基-2-羟基-3-(异丁胺基)丙基]氨甲酸叔丁酯(9) ; 9与7经磺化反应合成(2R,3S) -N-[(3-氨基-2-羟基-4-苯基)丁基]-N-异丁基-4-硝基苯磺酰胺盐酸盐-d4(10) ; 10依次经酰化、磷酰化、加氢还原和醋酸钙成盐反应合成了夫沙那韦-d4,总收率33%,纯度98.5%,其结构经1H NMR和EI-MS确证。
关键词:药物中间体;夫沙那韦-d4;合成
夫沙那韦钙盐[(1S,2R) -3-(4-氨基苯磺酰基) (2-甲基丙基)氨基-1-苯甲基-2-膦酰氧基-丙基-氨基甲酸(3S)四氢-3-呋喃酯钙盐(Ⅰ)]是由英国GSK和美国Vertex公司共同研发的高效蛋白酶抑制剂[1-2]。Ⅰ既可直接用于艾滋病治疗,也是合成安普那韦的前体药物[3-4]。Ⅰ水溶性良好,不良反应少[5-6]。
高纯度夫沙那韦-d4(14)可作为Ⅰ的内标,用于其生物利用率和药物代谢的研究[7]。目前,Ⅰ的相关合成工艺较为成熟,但14的合成报道相对较少。胡娟等[1]以L-苯丙氨酸为原料,经11步反应合成了Ⅰ,总收率10%。该路线反应时间长、产率低、后处理复杂。Emile等[8]以(2S,3S) -N-叔丁氧羰基-3-氨基-1,2-环氧-4-苯基丁烷为原料,成功合成了安普那韦,但未介绍羟基磷酰化的方法。Smiles等[9]以五氯化磷作磷酰化试剂,将对硝基苯磺酸转化为对硝基苯磺酰氯,产率高,操作简单。
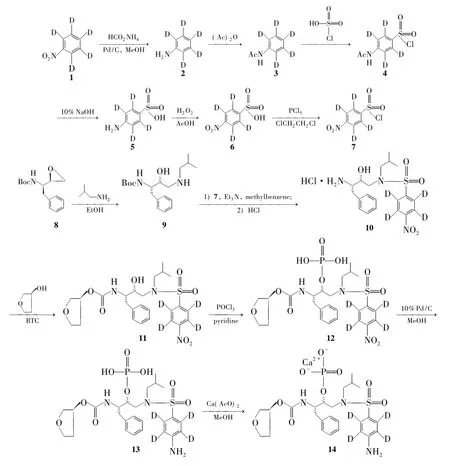
Scheme 1
在此基础上,本文设计了一条简便、经济、条件温和的合成14的反应路线。以硝基苯-d5(1)为原料,经还原、保护、磺酰化、脱保护、氧化和亲核取代反应制得中间体——对硝基苯磺酰氯-d4(7)。以(2R,3S) -N-叔丁氧羰基-3-氨基-1,2-环氧-4-苯基丁烷(8)为原料,经开环反应制得(1S,2R) -N-[1-苯甲基-2-羟基-3-(异丁胺基)丙基]氨甲酸叔丁酯(9) ; 9与7经磺化反应合成(2R,3S) -N-[(3-氨基-2-羟基-4-苯基)丁基]-N-异丁基-4-硝基苯磺酰胺盐酸盐-d4(10) ; 10依次经酰化、磷酰化、加氢还原和醋酸钙成盐反应合成了14(Scheme 1),总收率33%,纯度98.5%,其结构经1H NMR和EI-MS确证。
1 实验部分
1.1仪器与试剂
Buchi Melting Point B-545型熔点仪(温度未校正) ; Bruker-300 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标) ; Agilent 1100型高效液相色谱仪。
所用试剂均为分析纯。
1.2合成
(1)苯胺-d5(2)的合成
在反应瓶中加入1 12.0 g(0.1 mol)和MeOH 120 mL,搅拌使其溶解;氮气保护下加入甲酸铵30.7 g(0.49 mmol),于室温反应5 min。加入10% Pd/C 1.2 g,回流反应15 min。冷却至室温,抽滤,滤饼用甲醇洗涤,合并滤液和洗液,常压蒸馏得无色油状液体2 8.8 g,收率98.3 %。
(2)乙酰苯胺-d5(3)的合成
在反应瓶中加入2 8.8 g(89.8 mmol),冰浴冷却,氮气保护下加入乙酸酐20.2 g (197.8 mmol),搅拌下于室温反应2.5 h。冷却至室温,用饱和NaHCO3溶液(约50 mL)调至pH 7,用CH2Cl2(3×80 mL)萃取,合并萃取液,用饱和食盐水(150 mL)洗涤,无水Na2SO4干燥,蒸除溶剂得白色晶体3 12.5 g,收率99.2%,m.p.113℃~115℃。
(3)对乙酰胺基苯磺酰氯-d4(4)的合成
在反应瓶中加入3 6.0 g(42.9 mmol),冰盐浴冷却,氮气保护下滴加氯磺酸53.6 g (460 mmol),滴毕,于室温反应3 h[TLC检测,展开剂: V(乙酸乙酯)∶V(石油醚) = 1∶1]。冷却至15℃,搅拌下将反应液缓慢加入冰水中,析出大量白色固体,抽滤,滤饼用水(50 mL)洗涤,干燥得白色固体4 6.7 g,收率65.7%,m.p.147℃~149 ℃;1H NMR δ: 2.18(s,3H,CH3)。
(4)对氨基苯磺酸-d4(5)的合成
在反应瓶中加入4 13.0 g(57 mmol)和10% NaOH溶液300 mL,搅拌下于95℃反应3 h。冷却至室温,于15℃用10%HCl(约25 ml)调至pH 1~2,折出固体。抽滤,滤饼用5℃~8℃冷水(20 mL)洗涤,干燥得白色固体5 8.3 g,收率87.0%,m.p.279℃~281℃。
(5)对硝基苯磺酸-d4(6)的合成
在反应瓶中加入5 0.7 g(4 mmol)和冰醋酸5.6 mL,搅拌使其溶解;冰浴冷却,氮气保护下加入30% H2O25.6 mL,回流(80℃)反应4.5 h [TLC检测,展开剂: V (2%氯化氢的甲醇溶液)∶V(二氯甲烷) = 1∶2]。冷却至室温,蒸除溶剂,残余物干燥得黄色固体6(不纯化,直接投入下一步反应) 0.76 g,m.p.107℃~112℃; EIMS m/z(%) : 205.0(8),206.0(100),207.0 (8),208.0(5)。
(6) 7的合成
在反应瓶中加入6 0.5 g(2.4 mmol)和1,2-二氯乙烷7 mL,搅拌使其溶解;氮气保护下加入五氯化磷1.0 g(4.8 mmol),于65℃反应2.5 h。冷却至室温,蒸除溶剂,残余物干燥得黄色固体7 0.51 g,收率94.3%,m.p.75℃~80℃; EI-MS m/z(%) : 223.0(11),224.0(100),225.0(9),226.0(6)。
(7) 9的合成
在反应瓶中加入8 1.2 g(5 mmol,纯度>99 %)和无水乙醇15 mL,搅拌使其溶解;加入异丁胺6.6 mL(7.5 mmol),氮气保护下回流(78℃)反应1 h。冷却至室温,减压蒸除溶剂得白色固体9 1.53 g,收率99.8%。
(8) (2R,3S) -N-[(3-氨基-2-羟基-4-苯基)丁基]-N-异丁基-4-硝基苯磺酰胺盐酸盐-d4(10)的合成
在反应瓶中加入9 1.27 g(3.8 mmol)和干燥甲苯9 mL,搅拌使其溶解;氮气保护下于80℃滴加三乙胺0.42 g(4.2 mmol),滴毕,于90℃缓慢滴加7 1.0 g(4.5 mmol)的甲苯(4 mL)溶液,滴毕,反应2 h(TLC检测)。降温至80℃,缓慢滴加浓盐酸0.4 mL,滴毕,回流反应1 h。加入浓盐酸0.3 mL,反应0.5 h。蒸除溶剂,残余物加入乙酸乙酯6 mL,于60℃反应0.5 h。冰浴冷却,析出大量白色固体,抽滤,滤饼用冷乙酸乙酯(3 mL)洗涤,干燥得白色固体10 1.4 g,收率80.5%。
(9) (1S,2R) -3-(4-硝基苯磺酰基) (2-甲基丙基)氨基-1-苯甲基-2-羟基-丙基-氨基甲酸(3S)四氢-3-呋喃酯-d4(11)的合成
在反应瓶中加入三光气(BTC) 0.56 g(1.9 mmol)的二氯甲烷(15 mL)溶液,冰浴冷却至0℃~5℃;氮气保护下加入(S) -3-羟基四氢呋喃0.5 g(5.7 mmol)和三乙胺0.57 g(5.7 mmol),于室温反应3 h。减压蒸除溶剂,残余物用二氯甲烷(50 mL)溶解,加入10 2.3 g(5 mmol),于10℃滴加三乙胺1.4 g(14 mmol),于室温反应1 h;回流反应1 h(TLC检测)。减压蒸除溶剂,残余物加水30 mL,用二氯甲烷(3×25 mL)萃取,合并有机相,用饱和食盐水(10 mL)洗涤,无水硫酸钠干燥,抽滤,滤液蒸干后用混合溶剂[V(乙酸乙酯)∶V(乙醇) =4∶1,16 mL]重结晶得白色固体11 2.2 g,收率82.1%;H NMR δ: 7.25(m,5H,ArH),4.96~4.79(m,1H),4.11(m,1H),3.89~3.65(m,4H),3.49~3.3(m,2H),3.18~2.70(m,5H),2.60~2.49(m,1H),2.11~1.87 (m,3H),1.30~1.15(m,1H),0.94~0.85(m,6H,CH3)。
(10) (1S,2R) -3-(4-硝基苯磺酰基) (2-甲基丙基)氨基-1-苯甲基-2-膦酰氧基-丙基-氨基甲酸(3S)四氢-3-呋喃酯-d4(12)的合成
在反应瓶中加入11 1.0 g(1.9 mmol)和干燥吡啶50 mL,搅拌使其溶解;冰浴冷却至0℃~5℃,氮气保护下加入三氯氧磷1.72 g (11.2 mmol),于室温反应2.5 h(TLC检测)。冰浴冷却至5℃,加水20 mL,用甲基异丁基酮(3×30 mL)萃取,合并有机相,用饱和食盐水(25 mL)洗涤,无水硫酸钠干燥,抽滤,滤液蒸干后用甲基异丁基酮(50 mL)溶解,于室温滴加6 mol·L-1盐酸2 mL,滴毕,剧烈搅拌下于50℃反应2.5 h。冷却至室温,加水20 mL,静置分层,水相用甲基异丁基酮(2×30 mL)萃取,合并有机相,用饱和食盐水(20 mL)洗涤,无水硫酸钠干燥,抽滤,滤液蒸干得淡黄色固体12 1.0 g,收率84.7%。
(11) (1S,2R) -3-(4-氨基苯磺酰基) (2-甲基丙基)氨基-1-苯甲基-2-膦酰氧基-丙基-氨基甲酸(3S)四氢-3-呋喃酯-d4(13)的合成
在反应瓶中加入12 0.3 g(0.5 mmol)和甲醇7.5 mL,搅拌使其溶解;加入10%Pd/C 0.15 g,常压下通入氢气,于室温反应5 h[TLC检测,展开剂: V(二氯甲烷)∶V(甲醇) = 4∶1,醋酸2~3滴]。过滤,滤饼用甲醇(10 mL)洗涤,合并滤液和洗液,减压蒸除溶剂,剩余物干燥得白色固体13 0.27 g,收率91.5 %。
(12) 14的合成
在反应瓶中加入13 0.5 g(0.9 mmol)和甲醇10 mL,搅拌使其溶解;滴加一水合醋酸钙0.27 g (1.7 mmol)的水(2 mL)溶液,滴毕,于室温反应15 min。用5% NaOH溶液调至pH 8~9,析出大量白色固体,加水2 mL,搅拌10 min;抽滤,滤饼用50%乙醇洗涤,干燥得白色固体14 0.34 g,收率64.2%,纯度>98.5%,同位素丰度98.2%;1H NMR δ: 7.36~7.28(m,5H),4.96~4.79 (m,1H),4.49(m,1H),4.29~4.16(m,1H),3.89~3.55(m,4H),3.42~3.33(m,2H),3.16~2.90(m,3H),2.75~2.60 (m,1H), 2.11~1.85(m,2.5H),1.30~1.15(m,0.5H),0.85~0.79(m,6H) ; EI-MS m/z(%) : 587.1 (7),588.1(100),589.1(30),590.1(10)。波谱数据与文献[1]一致。
2 结果与讨论
首次以1原料,合成了高同位素丰度的7。用Pd/C-甲酸铵替代常规的Pd/C-H2还原硝基,反应更迅速,产率更高。氯磺酸与3发生亲电取代反应时应适当控制温度,避免因反应过于剧烈而导致氘原子丢失。用30% H2O2氧化氨基时,需用TLC即时监控,反应时间过长,副产物大量增加,难以纯化。
使用三光气取代羰基二咪唑进行酰化反应,大大缩短了反应时间,提高了产率。最后使用一水合醋酸钙水溶液成盐,只需调至pH为弱碱性,并多次重结晶达到高纯度和高同位素丰度。整个合成路线操作简便,所有步骤均只需重结晶提纯。
参考文献
[1]胡娟,肖元晶,韩峰燕,等.蛋白酶抑制剂Fosamprenavir Calcium的合成[J].中国医药工业杂志,2006,37(11) : 723-725.
[2]徐玉文,赵桂森.抗艾滋病药物研究进展[J].中国药物化学杂志,2002,12(2) : 119-124.
[3]张娜,赵宁,朱瑞恒,等.安普那韦的合成[J].合成化学,2008,16(1) : 115-117.
[4]唐红梅.抗艾滋病复制周期药物研究的新进展[J].广东医学,2005,26(9) : 1286-1288.
[5]刘晶,张娜,沈芸瑛,等.新型蛋白酶抑制剂的合成[J].合成化学,2008,16(5) : 593-595.
[6]谌志华,徐辉,庄璐,等.(2S,3S) -1,2-环氧基-3-叔丁氧酰胺基-4-苯丁烷的合成[J].合成化学,2013,21(3) : 361-363.
[7]Sorbera L A,Martin L,Castaner J,et al.Fosamprenavir[J].Drugs Future,2001,26(3) : 224-231.
[8]Al F E,Deininger D D,McGhie S,et al.Process for the synthesis of HIV protease inhibitor[P].WO 994 888 5,1999.
[9]Smiles S,Stewart J.p-Acetaminobenzenesulfonyl chloride[J].Org Syn Coll,1941,1: 8.
[10]Winkler R,Richter M E A,Knüpfer U,et al.Regioand chemoselective enzymatic N-oxygenation in vivo,in vitro and in flow[J].Angew Chem Int Ed,2006,45 (47) :8016-8018.
[11]Tung R D,Murcko M A.Sulfonamide inhibitors of HIV-asqartyl protease[P].WO 940 563 9,1994.
[12]Armitage I G,Searle A D,Singh H.Calcium (3S) -tetrahydro-3-furanyl(1S,2R) -3-{[(4-amino phenyl) sulfonyl](isobutyl) amino} -1-benzyl-2-(phosphonooxy) propylcarbamate[P].US 651 495 3B1,2003.
·综合评述·
Synthesis of High Purity(98.5%) Fosamprenavir-d4
SHI Lei1,SHEN Jia-lin1,SHEN Xiao-ming1,LÜAi-juan1,CHEN Li-qin2
(1.Nanjing Research Institute of Geology and Mineral Resources,Nanjing 210016,China; 2.Nanjing Jinglong Pharmaceutical Technology Co.,Ltd.,Nanjing 211800,China)
Abstract:An intermediate,p-nitrobenzenesulfonyl chloride-d4(7),was obtained by reduction,protection,sulfonylation,deprotection,oxidation and nucleophilic substitution,using nitrobenzene-d5as starting material.(1S,2R) -N-[1-benzyl-2-hydroxy-3-(isobutylamine) propyl]tert-butyl carbamate (9) was prepared by ring-opening reaction from (2R,3S) -N-Boc-3-amino-1,2-epoxy-4-phenylbutane.(2R,3S) -N-[(3-amino-2-hydroxy-4-phenyl) butyl]-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride-d4(10) was synthesized by sulfonation reaction of 9 with 7.Fosamprenavir-d4,in total yield of 33% and purity of 98.5%,was synthesized by acylation,phosphorylation,hydrogenation reduction and salification from 10,respectively.The structures were confirmed by1H NMR and EI-MS.
Keywords:drug intermediate; Fosamprenavir-d4; synthesis
作者简介:时磊(1983-),男,汉族,江苏南京人,硕士,主要从事药物合成和有机分析的研究。E-mail: njdkssl@163.com
*收稿日期:2014-10-30;
修订日期:2015-06-10
中图分类号:O625.75; R914.5
文献标识码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.07.0668
猜你喜欢
现代商贸工业(2016年14期)2016-12-27 15:35:49
安徽理工大学学报·自然科学版(2016年4期)2016-12-23 09:55:25
考试周刊(2016年85期)2016-11-11 02:09:06
农业与技术(2016年15期)2016-11-09 07:10:15
科技视界(2016年18期)2016-11-03 00:34:31
科教导刊·电子版(2016年19期)2016-08-19 18:05:29
中国市场(2016年28期)2016-07-15 04:18:49
科技视界(2016年15期)2016-06-30 00:46:57
科技视界(2016年10期)2016-04-26 15:31:06
科技视界(2016年9期)2016-04-26 09:47:50
