
HPLC-DAD 法同时测定真元颗粒干浸膏中5 个活性成分
2015-01-13 09:17李媛媛山金凤谭清杰蒋建兰
中成药 2015年9期
李媛媛, 山金凤, 谭清杰, 蒋建兰
(天津市生物与制药工程重点实验室,天津大学化工学院制药工程系,天津300072)
真元颗粒是由刺五加、青蒿、葛根、防风、甘草等九味中药经提取加工制成的复方制剂。在临床上用于辅助肿瘤治疗,达到改善肿瘤患者体质、提高机体免疫、提高生存质量、延长寿命的效果[1]。该制剂已建立了以甘草酸为指标成分进行定量测定的质量控制标准,考虑到中药复方制剂成分的多样性和复杂性,单一成分作为指标不能准确反映中药复方制剂质量,因而本实验建立了一种同时测定真元颗粒干浸膏中刺五加和青蒿有效成分绿原酸[2-3]、葛根有效成分葛根素[4]、防风有效成分升麻素苷[5]、甘草有效成分甘草苷和甘草酸的高效液相色谱法[6]。
1 仪器与试药
1.1 仪器 Agilent 1260 Infinity 型高效液相色谱仪,配四元梯度泵600 bar,超高灵敏度二极管阵列检测器,半导体控温柱温箱,100 位自动进样器,Chemstation 工作站(美国Agilent 公司);电子天平BSA224S-CW 购自赛多利斯科学仪器(北京)有限公司;AS10200 超声波清洗机(电压220 V/50 Hz)购自天津奥特赛恩斯仪器有限公司;Eppendorf centrifuge 5804R 型离心机。
1.2 试药 甘草苷、甘草酸铵、升麻素苷、葛根素、绿原酸对照品(批号分别为111610-201106、110731-201317、 111522-201310、 110752-201313、110753-201314)均购自中国食品药品检定研究院;真元颗粒中试干浸膏,中新制药厂生产;中药复方药材(安仁中医院)。提取用蒸馏水,天津大学自制;无水乙醇为分析纯;磷酸为优级纯;冰醋酸,甲酸,甲醇和乙腈为色谱纯;娃哈哈纯净水。药材由天津药物研究院张铁军研究员鉴定。
2 方法与结果
2.1 色谱条件 Agilent SB C18色谱柱(250 mm ×4.6 mm,5 μm);流动相为乙腈(A)-0.1%甲酸溶液(B),梯度洗脱(0 ~5 min,95% B;5 ~43.5 min,95% ~88.5% B;43.5 ~66.5 min,88.5% ~83% B;66.5 ~79 min,83% ~80% B;79 ~80 min,80% ~69%B;80 ~104 min,69% ~58%B);柱温30 ℃;体积流量1.2 mL/min;检测波长为250 nm (0 ~43.5 min),300 nm (43.5 ~66.5 min),250 nm (66.5 ~104 min);进样量为10 μL。在上述条件下,样品中各对照品与其他组分达到基线分离,与相邻的色谱峰分离度大于1.5。各对照品峰理论塔板数不小于16 000。
2.2 对照品溶液的配制 分别精密称取干燥至恒定质量的对照品绿原酸、葛根素、升麻素苷、甘草苷和甘草酸铵 (甘草酸质量= 甘草酸铵质量/1.020 7)适量,加甲醇制成质量浓度分别为916、780、664、1 900 μg/mL 和900 μg/mL 的各对照品溶液,4 ℃冷藏备用。
2.3 供试品溶液的制备 精密称取真元颗粒干浸膏粉末0.200 0 g,置5 mL 量瓶中,加入40%甲醇,密塞,超声(40 kHz、300 W)处理30 min,放冷至室温,40%甲醇定容至刻度,摇匀,滤过,过0.45 μm 微孔有机滤膜,取续滤液,即得。
2.4 阴性供试品溶液的制备 按真元颗粒处方称取药材,按“2.3”项下方法分别制备缺刺五加和青蒿、缺葛根、缺甘草和缺防风的阴性供试品溶液,4 ℃冷藏备用。
2.5 方法学考察
2.5.1 方法专属性试验 在“2.1”项色谱条件下,所测5 种有效成分与其他成分色谱峰均可基线分离(分离度大于1.5),且阴性供试品无干扰。对照品、供试品及阴性供试品色谱图见图1。
2.5.2 线性关系的考察 分别精密吸取“2.2”项下各对照品溶液适量,混合定容至25 mL 量瓶中,制成含绿原酸、葛根素、升麻素苷、甘草苷和甘草酸铵质量浓度分别为82.4、46.8、19.9、266.0 和576.0 μg/mL 的混合对照品溶液(1)。精密吸取混合对照品溶液(1)3 800、2 500、1 940、1 250、625 μL 分别置于5 mL 量瓶中,加甲醇定容后摇匀,制得混合对照品溶液(2) ~ (6)。取混合对照品溶液(1) ~(6),按“2.1”项色谱条件记录色谱图及峰面积。以峰面积为纵坐标(y),质量浓度(μg/mL)为横坐标(x),绘制标准曲线,计算得回归方程绿原酸y=10.547x+4.293 4 (r=0.999 6);葛根素y=35.382x +19.105 (r =0.999 4);升麻素苷y =12.346x +3.931 9 (r =0.999 7);甘草苷y =8.650 9x +18.898 (r =0.999 6);甘草酸铵y=6.759 6x -72.048 (r =0.999 3)。绿原酸、葛根素、升麻素苷、甘草苷和甘草酸铵分别在10.3 ~82.4 μg/mL、5.9 ~46.8 μg/mL、2.5 ~19.9 μg/mL、33.3 ~ 266.0 μg/mL、72.0 ~ 385.5 μg/mL范围内具有良好线性关系。
2.5.3 精密度试验 取混合对照品溶液(3),按“2.1”项下色谱条件连续进样6 次,绿原酸、葛根素、升麻素苷、甘草苷和甘草酸铵峰面积的RSD 值分别为0.85%、0.88%、0.85%、0.78%和0.67%。结果表明,仪器精密度良好。

图1 HPLC 色谱图Fig.1 HPLC chromatograms
2.5.4 供试品精密度试验 取真元颗粒干浸膏粉,按“2.3”项下方法制备供试品溶液,按“2.1”项下色谱条件连续进样6 次,绿原酸、葛根素、升麻素苷、甘草苷和甘草酸铵峰面积的RSD 值分别为0.53%、0.75%、1.58%、0.17% 和0.13%。结果表明,供试品溶液制备方法适合甘草酸等5 个成分的HPLC 测定。
2.5.5 重复性试验 取同一批号真元颗粒干浸膏粉,按“2.3”项下方法制备供试品溶液6 份,分别按“2.1”项下色谱条件测定。绿原酸、葛根素、升麻素苷、甘草苷和甘草酸峰面积的RSD 值分别为1.2%、0.57%、0.71%、1.69%、0.72%。结果表明,方法重复性良好。
2.5.6 稳定性试验 取真元颗粒干浸膏粉,按“2.3”项下方法制备供试品液,于0、2、4、6、12、24 h 按“2.1”项下色谱条件测定。绿原酸、葛根素、升麻素苷、甘草苷和甘草酸5 个化合物峰面积RSD 分别为1.90%、0.74%、1.30%、0.20%和0.25%。结果表明,在24 h 内供试品溶液基本稳定。
2.5.7 加样回收率试验 精密吸取“2.2”项下各对照品溶液适量,混合定容至25 mL 量瓶中,制成含绿原酸、葛根素、升麻素苷、甘草苷和甘草酸铵分别为48.7、25.2、10.6、130.5 和316.8 μg/mL的混合对照品溶液(7)。取含有量已知的真元颗粒干浸膏约0.1 g,精密称定,平行制备9 份,每3 份为一组,按高、中、低 (即80%、100%、120%)比例分别精密吸取加入混合对照品溶液(7),按“2.3”项下方法制备供试品液,按“2.1”项下色谱条件测定,计算回收率,结果见表1。
2.6 样品测定 取3 批不同来源药材制备的样品,按“2.3”项下方法制备供试品液,按“2.1”项下色谱条件测定,结果见表2。
3 讨论
3.1 供试品溶液制备方法考察 实验中对提取溶剂和超声时间进行了考察。提取溶剂考察了甲醇、乙醇、80%甲醇、40%甲醇、80%乙醇和40%乙醇,结果表明40%甲醇配制的供试品溶液,谱峰分离效果最好,色谱峰响应更大,故确定采用40%甲醇配制供试品溶液。超声时间考察了30、40、50 和60 min 不同时间的提取效果,结果表明在超声提取30 min 后绿原酸、葛根素、升麻素苷、甘草苷和甘草酸的量无显著变化,故选取超声30 min。

表1 加样回收率试验结果Tab.1 Result of recovery tests
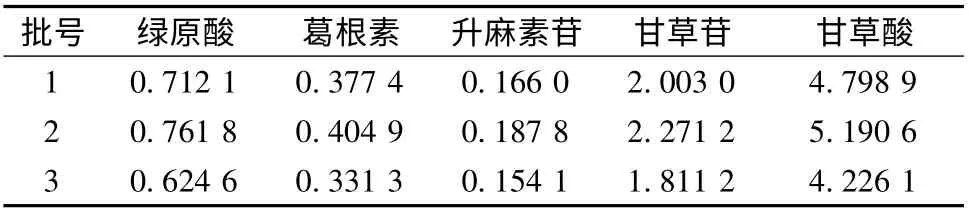
表2 样品测定结果(mg/g)Tab.2 Result of determination of samples(mg/g)
3.2 检测波长的选择 设定237、250、280 和300 nm 四个波长进行筛选,同时利用高效液相色谱仪中DAD 检测器的波长扫描功能,进行190 ~400 nm 全波长扫描,得到绿原酸、葛根素、升麻素苷、甘草苷和甘草酸的紫外吸收光谱。综合来看,250 nm 下各色谱峰均有较大吸收,其中绿原酸峰虽然在320 nm 左右有最大吸收,但因其含有量相对较高,在250 nm 下色谱峰响应已较大,故选定250 nm 为绿原酸的检测波长;其中升麻素苷和甘草苷色谱峰在250 nm 条件下由于杂峰干扰,分离度太差,经多次梯度改变尝试仍无果,因而考虑在升麻素苷和甘草苷均有较大吸收且效果与250 nm 极其相近的300 nm 下进行检测。故本实验利用波长切换,更加有效的进行了多成分定量测定。
3.3 流动相的选择 参考相关文献[7-13],本实验对乙腈-0.1%醋酸溶液、乙腈-0.1%磷酸溶液和乙腈-0.1%甲酸溶液进行了比较,结果发现乙腈-0.1% 甲酸溶液梯度洗脱时,各色谱峰保持良好的峰形,分离度均大于1.5,理论板数均超过了10 000,并且阴性对照色谱中无干扰峰的出现。故确定流动相组成为乙腈-0.1%甲酸溶液。
3.4 柱温和体积流量的选择 本实验考察25 ℃、30 ℃、35 ℃这3 个不同柱温的影响,结果表明25℃和35 ℃下色谱峰分离效果较差,而30 ℃柱温色谱图中各色谱峰峰形良好,分离度均符合要求,故确定最佳柱温为30 ℃。试验考察了0.8、1.0、1.2 mL/min 这3 个不同体积流量的影响,结果表明体积流量为0.8 mL/min 和1.0 mL/min 时,部分色谱峰分离效果差,而体积流量为1.2 mL/min时各目标色谱峰的峰形良好,分离度均达到要求,故确定体积流量为1.2 mL/min。
3.5 结论 本实验建立HPLC-DAD 同时定量测定真元颗粒干浸膏中绿原酸、葛根素、升麻素苷、甘草苷和甘草酸的方法,针对5 个活性指标成分进行分析,简便易行,专属性强,重复性好,准确度高,为真元颗粒质量控制提供了科学依据。
[1] 张晓杭,袁 乐,李媛媛,等. 真元颗粒质量标准的研究[J]. 中药材,2014,37(8):1478-1482.
[2] 龚婧如,王书芳. HPLC 多波长法测定刺五加颗粒中5 个化合物的含量[J]. 药物分析杂志,2013,33 (4):595-598.
[3] 邱华荣,田 吉,冯文宇,等. 青银注射液中绿原酸与樟脑的含量测定[J]. 中成药,2004,26(8):621-623.
[4] 陈小新,赖小平,李 耿,等. 葛根素自微乳在大鼠体内的药代动力学研究[J]. 中成药,2011,33 (7):1220-1222.
[5] 黄 澜,陈惠玲,李玲玲. HPLC 同时测定正天丸中芍药苷、阿魏酸、升麻素苷、5-O-甲基维斯阿米醇苷的含量[J]. 中国中药杂志,2013,38(13):2114-2117.
[6] 陈云华,赵晓霞,王文全,等. 高效液相色谱法同时测定甘草中甘草酸、甘草苷、异甘草素的含量[J]. 中国中医药信息杂志,2009,16(8):52-54.
[7] 段天璇,于密密,刘春生,等. HPLC 法同时测定甘草指纹图谱暨甘草苷、甘草酸含量[J]. 中成药,2006,28(2):161-165.
[8] 黄惠莲,周燕双,宋丽莉. 不同提取工艺下甘草提取物中甘草酸铵含量测定研究[J]. 中国当代医药,2011,18(33):47-50.
[9] 陈新阳,毕开顺,易丽昕,等. 双波长切换HPLC 同时测定桂枝甘草汤中4 种成分的含量[J]. 中国现代应用药学,2011,28(3):265-267.
[10] 王欣之,文红梅,李 伟,等. HPLC 法同时测定玉屏风散中色原酮、黄酮和内酯类9 种成分的含量[J]. 中药新药与临床药理,2009,20(2):142-146.
[11] 毕何锋,张 婕,蒋东旭,等. HPLC 法测定柴葛解肌汤中葛根素旳含量[J]. 中国药师,2014,17(1):161-162.
[12] 于风平,连传宝. 超快速液相色谱法测定刺五加注射液中紫丁香苷和绿原酸的含量[J]. 中国药师,2010,13(12):1769-1770.
[13] 孙玉刚,秦续文,张 玲,等. 高效液相色谱法同时测定青蒿绿原酸、隐绿原酸、东莨菪内酯含量[J]. 天津中医药,2012,29(5):484-486.
猜你喜欢
中成药(2022年7期)2022-12-02
昆明医科大学学报(2021年4期)2021-07-23
中国现代中药(2020年8期)2020-10-28
中国中医基础医学杂志(2020年7期)2020-01-13
中成药(2019年12期)2020-01-04
中成药(2018年6期)2018-07-11
中成药(2018年5期)2018-06-06
中成药(2017年6期)2017-06-13
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
中国当代医药(2015年33期)2015-03-01
