
基于支持向量机构象性B 细胞表位预测研究
2015-01-12 02:44张春华
吉林大学学报(信息科学版) 2015年3期
刘 波,张春华
(东北师范大学a.体育学院,长春130024;b.计算机科学与信息技术学院,长春130117)
0 引言
在体液免疫过程中,B细胞表面受体(BCR)或抗体识别外源抗原蛋白,并产生免疫应答,这部分抗原表面区域被称为B细胞表位。准确定位B细胞表位是现代免疫学和疫苗学研究的关键。B细胞表位预测的研究目标是模拟天然表位结构和功能的分子在医学诊断和治疗以及疫苗设计中取代天然表位[1]。定位表位可靠的方法是通过生物实验方法[2,3],例如X射线衍射、核磁共振、定点突变和缺失及蛋白质印记等,但实验方法费时费力且成本很高。因此,首先通过计算的方法预测出可能的候选表位,再进一步做实验验证,能在很大程度上节约成本,提高工作效率。
从空间构象上区分,B细胞表位可分为线性表位和构象性表位[4]。线性表位是抗原分子表面肽链上连续的氨基酸序列,而构象性表位是抗原表面不同的氨基酸序列片段通过空间折叠聚集形成的一个表面区域。研究表明,B细胞表位只有一少部分是线性的,90%都是构象性表位[1,4]。
理论上,蛋白质3D结构可比一级序列提供更多信息,因此,利用蛋白质3D结构预测表位能得到更好的结果[5,6]。Kolaskar等[7]以蛋白质3D结构为前提,通过计算抗原蛋白氨基酸的表面可及性,分析和定位日本脑炎病毒的构象性表位。为进一步完善算法,在2005年发布了第1个基于Web的构象性B细胞表位预测软件CEP(Conformational Epitope Prediction)[8]。随后,多个基于蛋白质3D结构的B细胞表位预测方法被提出[9-16],表1给出了基于结构的B细胞表位预测方法及对应的网址,介绍了每种方法使用的特征和分类方法。
尽管这些方法的性能都具有不同程度的提高,但由于表位是背景独立的,所以表位预测仍然是个具有挑战性的难题[17]。

表1 基于结构的B细胞表位预测方法Tab.1 B-cell epitope prediction methods based on antigen structures
在提取特征方面,上述方法提取了抗原的单一结构特征或几个结构特征的组合;在预测方法方面,这些方法或直接使用结构特征的得分,或使用投票机制。文献[13]和文献[16]使用了两种不同的机器学习方法,文献[13]提取了蛋白质残基的理化属性和几何特征,并使用SVM(Support Vector Machine)对残基进行分类,进而得到候选表面氨基酸;文献[16]使用RF(Randon Forest)对基于蛋白质氨基酸的距离特征进行表位预测。从预测性能看,他们的预测效果较好,比其他方法有一定的提高。B细胞标为预测方法所选用的特征直接影响了预测方法的结构,从以上几种预测方法的结果看,目前所利用的特征以及特征组合不足以完全表征B细胞表位的全部特征,并将其与非表位残基区分。
笔者在上述研究的基础上,提取了基于抗原结构的10个表位相关属性特征,这些特征反映了残基的表位倾向性、残基保守性、残基无序性,抗原表面的紧凑程度、可平面性、二级结构组成和原子能量,最后使用LibSVM对抗原表面氨基酸进行分类[18]。通过几个测试例,验证笔者算法的有效性,并与以上可用的、有返回预测结果的其他基于结构的B细胞表位预测的方法预测结果进行了比较,结果表明,笔者算法的精确度最高。
1 基于支持向量机的B细胞表位预测方法
1.1 表位特征提取
基于抗原结构的B细胞表位预测就是基于不同的表位特征对抗原表面氨基酸进行分类,如何选取有效B细胞表位度量属性特征对预测算法的性能至关重要。笔者在基于抗原结构的B细胞表位预测方法相关研究的基础上,选取了以下10个有效的B细胞表位度量属性特征。
1)残基表位倾向性。残基表位倾向性反映的是氨基酸残基在某种意义上是表位的概率,可通过公式[13]

表2 20个氨基酸的值Tab.2 Thevalue of 20 amino acids

表2 20个氨基酸的值Tab.2 Thevalue of 20 amino acids
ln(Pinterfacer /ln(Pinterfacer /氨基酸Psurfacer )/Saver氨基酸Psurfacer )/Saver ln(Pinterfacer /ln(Pinterfacer /氨基酸Psurfacer )/Saver氨基酸Psurface r )/Saver Ala -0.392 Leu -1.31 Gln -0.006 Trp -0.07 Arg 0.316 Lys 0.021 Glu -0.492 Val -0.826 Asn 0.446 Met 1.06 Gly 0.463 Ser -0.004 Asp -0.307 Phe 0.979 His 0.207 Thr -0.062 Cys -7.36 Pro 0.017 Ile 0.334 Tyr 0.979
2)残基保守性得分。在一般情况下,抗原-抗体相互作用表面的残基保守性较低,所以残基保守性得分越低的残基越有可能是表位残基。残基的进化保守性得分是基于多序列比对和系统发生树计算得到的,笔者使用Rate4Site计算残基的保守性得分[19]。
3)无序得分。蛋白质片段在生理条件下缺乏固定的三维空间结构,在生物学功能中有重要作用。蛋白质的无序区域考虑到更多的修饰位点和相互作用对,并且通常包含PTM(Protein Post-Translational Modification)位点,类别信号和蛋白质配体。因此,这些区域对蛋白质结构和功能相当重要,笔者通过VSL2提取蛋白质氨基酸残基的无序得分[20]。
4)接触数目。接触数目反映残基所在蛋白质表面区域的氨基酸紧凑程度,一般认为接触数量越小,越有可能是表位。本文中残基的原子接触数目的计算公式如下
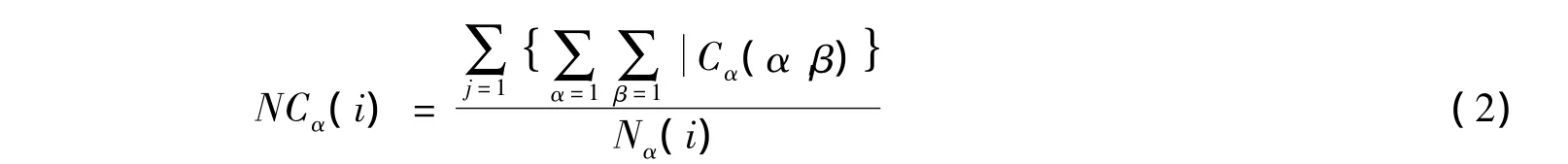
其中

其中Cα为两个原子之间的接触,Nα(i)为残基中原子的数目。如果两个原子之间的距离在0.5 nm[21-23],则认为它们是接触的。
5)可平面性得分。可平面性得分反映蛋白质表面区域的突出程度。蛋白质表面的几何属性可以影响蛋白质相互作用。笔者通过突出指数和深度指数衡量氨基酸残基的可平面性。其中,突出指数定义为蛋白质球体内部体积与剩余部分体积之比的倒数;深度指数定义为离它最近的溶剂可及原子的距离,对于溶剂可及性原子,其深度指数为0,对于掩埋于蛋白质内部的原子,其深度指数大于0,掩埋越深的原子,其深度指数的值越高。笔者采用PSAIA1计算原子的突出指数和深度指数[24]。
6)二级结构组成。二级结构组成也是基于patch的特征。有研究表明抗原的二级结构组成中转角和环结构与B细胞表位的关联性较强[24]。笔者用“H”表示‘helix’螺旋结构,用“E”表示‘strand’折叠结构,用“C”表示‘other’卷曲结构,使用SSPRO8计算氨基酸残基对应的三种结构[25],并分别用100,010和001对3种情况进行编码。
7)原子溶剂化能。溶剂化作用是溶剂分子通过它们与离子的相互作用,而累积在离子周围的过程。该过程形成离子与溶剂分子的络合物,并放出大量的热。溶剂化作用改变了溶剂和离子的结构。溶剂化作用也是高分子和溶剂分子上的基团能相互吸引,从而促进聚合物的溶解,原子的溶剂化能反映了复合物的生成和相互作用。原子溶剂化能的计算公式如下

其中σ(i)是原子i的能量权重;α(i)为原子i的溶液排除表面积,α(i)通过MSMS计算[26]。原子融化参数如表3所示。
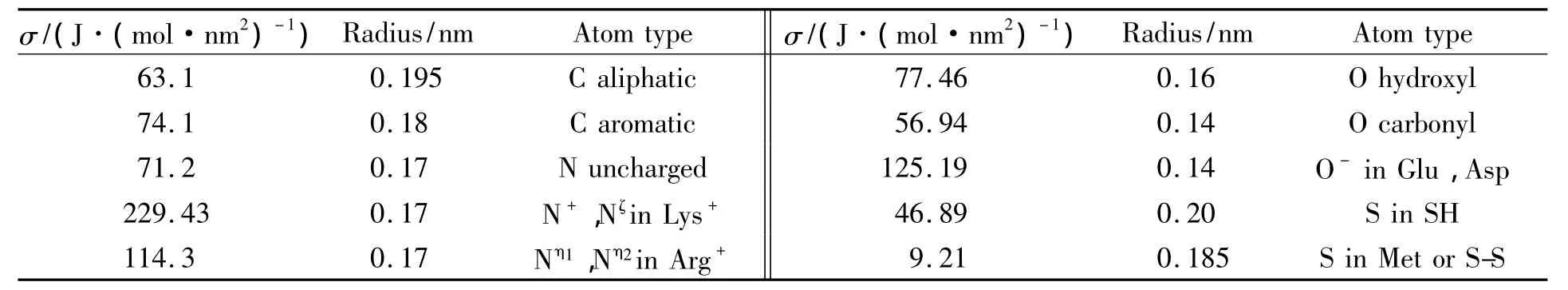
表3 原子融化参数Tab.3 The atom solvent parameter
笔者共提取10个抗原结构相关特征,其中残基表位倾向性1个,残基保守性得分1个,无序得分1个,接触数目1个,可平面性得分2个,二级结构组成3个,原子溶剂化能1个。
1.2 基于支持向量机的抗原表面氨基酸分类
提取了氨基酸的10个表位属性特征后,笔者使用LibSVM对抗原表面的氨基酸进行分类。为了提高基于机器学习方法的分类预测性能,正负样本的比例最好均衡为1∶1左右。但经过统计分析,笔者所解决的问题中,表位残基与非表位残基的比例大约为1∶10,而传统的解决方法是补偿正样本和对负样本进行采样,然而这样的方式显然不适合解决笔者的问题。因此,笔者采用为正样本设置权重的处理技巧均衡样本。同时,为了提高支持向量机的训练速度,笔者采用了对数据进行分块[27]的处理手段。
1.3 性能评价参数
笔者采用下面几个评价参数对算法的性能进行分析:敏感性、特异性和精确度,计算公式如下

其中TP为正确预测的表位残基数目;FP为错误预测的表位残基数目;TN为正确预测的非表位残基数目;FN为错误预测的非表位残基数目。除此之外,笔者还用AUC1评价算法的测试性能。AUC是指敏感性/1-特异性的曲线下面积。其中,敏感性是指被正确预测的残基数目占测试集中所有残基数目的比例,它衡量了方法正确预测表位残基的能力。特异性是指正确预测的非表位残基数目占总体非表位残基数目的百分比,它衡量了方法正确预测的非表位残基的能力。精确度是指正确识别的表位残基数目占所有预测候选表位残基数目的比例。ROC曲线是用构图法揭示敏感性和特异性的相互关系,它通过将连续变量设定多个不同的临界值,从而计算出一系列敏感性和特异性,再以敏感性为纵坐标、1-特异性为横坐标绘制成曲线,AUC即为ROC曲线下面积,面积越大,表明预测的准确性越高。
2 结果与分析
2.1 数据集
笔者的训练集来自3部分:1)2007年在华盛顿召开的首届针对B细胞表位预测软件评价的研讨会上公布的标准测试集[28];2)protein-protein docking的标准测试集2.0版[29];3)相关论文中用到的测试例[9-16]。删除3个部分中的重复数据,并删除包含超过一条抗原链的结构,最后得到161个无重复且含有一条抗原链的复合体结构作为笔者的测试集。B细胞表位定义为抗原表面与抗体残基相互作用表面积小于等于0.04 nm2的氨基酸残基。笔者的测试集选自Mayrose在Epitopia中使用的数据集[13],删除其中具有2条抗原链的结构,最后确定15个结构为笔者的测试集。
2.2 不同机器学习方法及参数对算法的影响
在笔者的数据集中,负样本的数量明显高于正样本,为了在不平衡的样本上得到最优的预测结果,笔者除了使用SVM分类方法外,还选用了RF分类方法。笔者的LibSVM和RF分类方法是在Weka 3.7下建立的接口,并构造分类器。除了两种方法的默认参数外,笔者还测试了其他的参数组合,表4列出了几种代表参数组合下的结果。

表4 不同机器学习方法及参数下算法的结果Tab.4 Prediction results of different machine learning methods and parameters
从表4可以看出,在笔者的数据集上,SVM分类方法的预测结果略优于随机森林方法;对于SVM方法,采用为正样本加权处理不平衡样本的方式略优于分块处理方式,而采用二者结合的方式得到的预测结果最优。
2.3 算法参数设定
在使用SVM进行分类时,笔者为正样本进行加权进而均衡训练样本。笔者对权重值从3~10,步长0.1进行了测试,表5给出了权重值分别为6.5、7和7.5时以及使用和不使用分快处理技术时算法在训练集上的预测敏感性、特异性、精确度和AUC(Area Under roc Curve)。

表5 算法在不同权重值下的预测性能比较Tab.5 Prediction results with different weights
通过表5可以看出,在权重值为7.5时,算法的整体预测性能最高。同时,对数据进行分块处理后,虽然预测性能没有提高,但网络的训练速度明显提高。因此,笔者算法使用支持向量机的分类方法,为正样本加权7.5,并对数据采用分块处理。
2.4 算法测试结果分析
表6给出了笔者算法的测试结果,抗原大小为抗原所包含的氨基酸数目,表位大小为抗原表位残基数目。
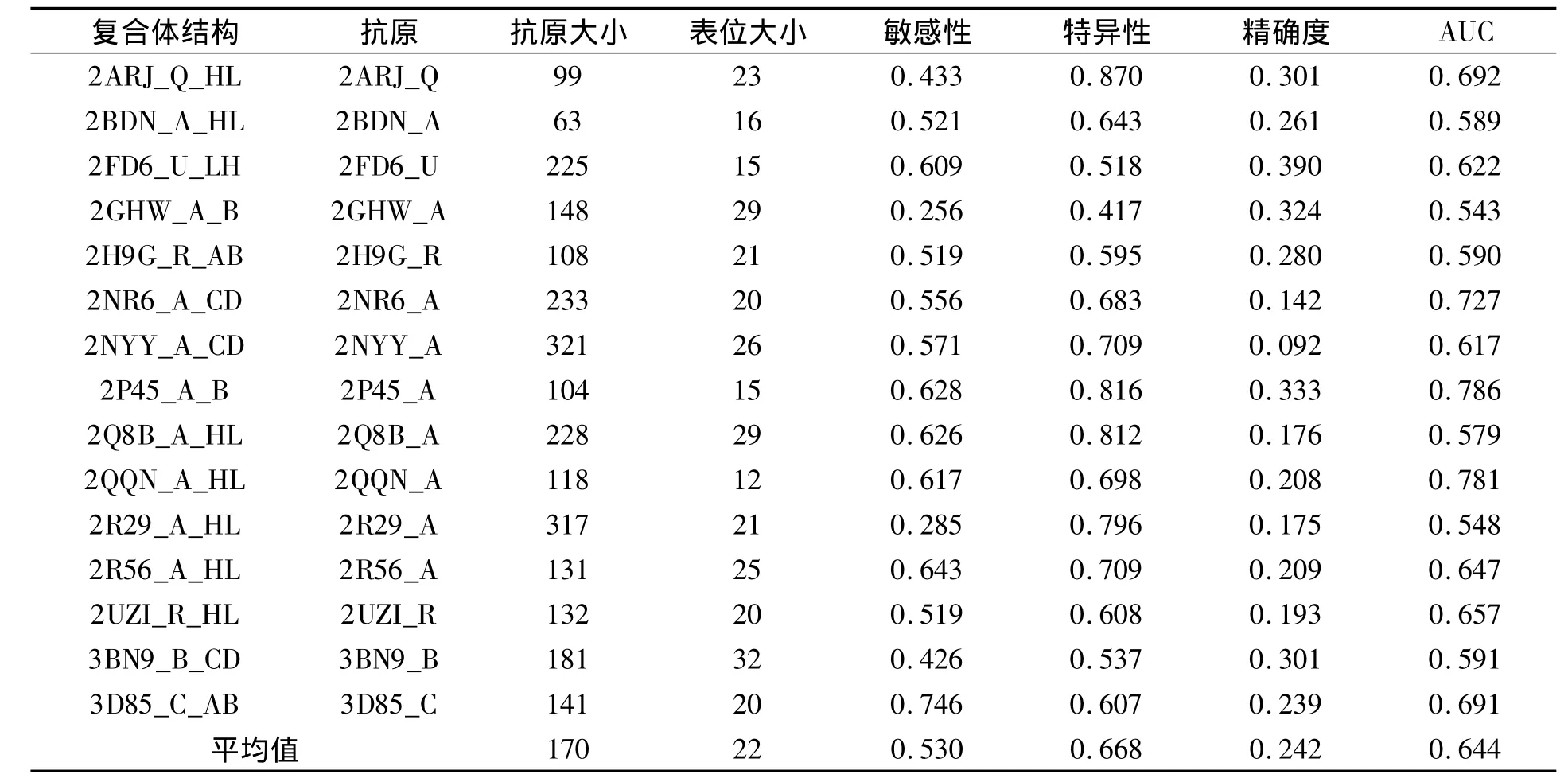
表6 笔者算法的测试结果Tab.6 Prediction results of our method
从表6可以看出,对于大部分数据,笔者的算法都能较准确预测表位所在区域。图1给出了笔者算法预测结果的敏感性和1-特异性散点图,可以看出,除2GHW_A和3BN9_B这2条数据外,所有测试例的结果都在对角线上,预测结果验证了笔者算法的有效性。
2.5 与其他算法的结果比较
为了进一步验证笔者算法的有效性,笔者使用独立测试集测试了几个其他可用的基于结构的构象性B细胞表位预测方法,预测结果如表7所示。

图1 算法的敏感性/1-特异性散点图Fig.1 The scatter diagram of sensitivity and 1-specificity
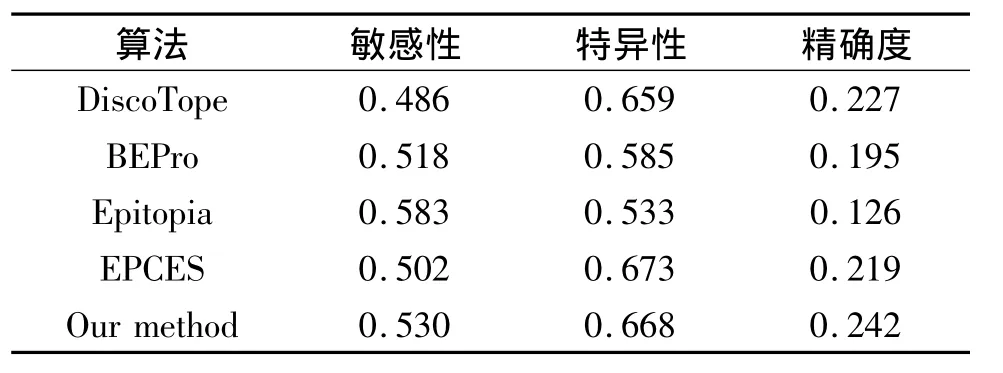
表7 笔者算法的预测结果与其他几种算法的比较Tab.7 The prediction results compared with other methods
表7给出了笔者方法和其他4种基于结构的构象性B细胞表位预测方法在笔者独立测试集上的平均预测性能比较。从表7可以看出,在笔者的测试集上,Epitopia的敏感性最高,达到了58.3%,笔者方法在测试集上的平均敏感性略低;EPCES的特异性最高,达到了67.3%,笔者的方法次之,为66.8%;从精确度看,笔者的方法最高,达到了24.2%。
笔者选取了15个复合体结构的绑定结构作为算法的测试例,验证算法的有效性。结果表明:算法的预测性能较稳定,个别数据预测结果差异较大,最好的预测结果精确度达到39%,有一条数据的预测结果相对较差,精确度仅有9%。这种差异说明笔者所提取的氨基酸表位属性特征可以对抗原表面氨基酸进行大致的分类,但这些特征的特异性是不全面的,不能进行细致全面精确地分类。因此,要提高算法的预测性能,必须挖掘更全面更有效的B细胞表位属性特征以及属性特征的组合。
3 结 语
笔者研究了基于抗原结构的B细胞表位预测,通过提取10个B细胞相关表位的结构属性特性,并使用SVM对抗原表面氨基酸进行分类,从而实现对抗原表面B细胞表位的预测。从预测方法的平均性能看,笔者方法的敏感性为0.53,特异性为0.668,精确度为0.242,AUC为0.644。在今后的工作中,将重点研究如何提取更加有效的B细胞表位属性特征以及优化算法的参数,提高预测方法的整体性能。
[1]IRVING M B,PAN O,SCOTT J K.Random-Peptide Libraries and Antigen-Fragment Libraries for Epitope Mapping and the Development of Vaccines and Diagnostics[J].Curr Opin Chem Biol,2001,5(3):314-324.
[2]RUS J J,BURNETT R M.Type-Specific Epitope Locations Revealed by X-Ray Crystallographic Study of Adenovirus Type 5 Hexon [J].Mol Ther,2000,1(1):18-30.
[3]MAYER M,MEYER B.Group Epitope Mapping by Saturation Transfer Difference NMR to Identify Segments of a Ligand in DirectContact with a Protein Receptor[J].J Am Chem Soc,2001,123(25):6108-6117.
[4]REGENMORTEL M H VAN.AntigenicityandImmunogenicityofSyntheticPeptides [J].Biologicals, 2001,29(3/4):209-213.
[5]SUN Pingping,CHEN Wenhan,HU Yanxin,et al.Epitope Prediction Based on Random Peptide Library Screening:Benchmark Dataset and Prediction Tools Evaluation [J].Molecules,2011,16(6):4971-4993.
[6]THORNTON J M,EDWARDS M S,TAYLOR W R,et al.Location of Continuous'Antigenic Determinants in the Protruding Regions of Proteins[J].Embo J,1986,5(2):409-413.
[7]KOLASKAR A S,KULKARNI-KALE U.Prediction of Three-Dimensional Structure and Mapping of Conformational Epitopes of Envelope Glycoprotein of Japanese Encephalitis Virus[J].Virology,1999,261(1):31-42.
[8]KULKARNI-KALE U,BHOSLE S,KOLASKAR A S.CEP:A Conformational Epitope Prediction Server[J].Nucleic Acids Res,2005,33(Web Server Issue):168-171.
[9]HASTE ANDERSEN P,NIELSEN M,LUND O.Prediction of Residues in Discontinuous B Cell Epitopes Using Protein 3D Structures[J].Pernille Haste Andersen,Morten Nielsen and Ole Lund Protein Science,2006,15(11):2558-2567.
[10]PONOMARENKO J,BUI H H,LI W,et al.ElliPro:A New Structure-Based Tool for the Prediction of Antibody Epitopes[J].BMC Bioinformatics,2008,9(2):514.
[11]MICHAEL J.SWEREDOSKI,PIERRE BALDI.PEPITO:Improved Discontinuous B-Cell Epitope Prediction Using Multiple Distance Thresholds and Half Sphere Exposure[J].Bioinformatics,2008,24(12):1459-1460.
[12]SUN Jing,WU Di.SEPPA:A Computational Server for Spatial Epitope Prediction of Protein Antigens[J].Nucl Acids Res,2009,37(S2):612-616.
[13]RUBINSTEIN N D,MAYROSE I,MARTZ E,et al.Epitopia:a Web-Server for Predicting B-Cell Epitopes [J].BMC Bioinformatics,2009,14(10):287.
[14]LIANG S,ZHENG D,ZHANG C,et al.Prediction of Antigenic Epitopes on Protein Surfaces by Consensus Scoring[J].BMC Bioinformatics,2009,22(10):302.
[15]LIANG S,ZHENG D,STANDLEY D M,et al.EPSVR and EPMeta:Prediction of Antigenic Epitopes Using Support Vector Regression and Multiple Server Results[J].BMC Bioinformatics,2010,16(11):381.
[16]ZHANG Wen,XIONG Yi,ZHAO Meng,et al.Prediction of Conformational B-Cell Epitopes from 3D Structures by Random Forests with a Distance-Based Feature[J].BMC Bioinformatics,2011,17(12):341.
[17]GREENBAUM J A,ANDERSEN P H,BLYTHE M,et al.Towards a Consensus on Datasets and Evaluation Metrics for Developing B-Cell Epitope Prediction Tools[J].J Mol Recognit,2007,20(2):75-82.
[18]CHANG C C,LIN C J.LIBSVM:A Library for Support Vector Machines[J].ACM Transactions on Intelligent Systems and Technology,2011,27(2):1-27.
[19]ITAY MAYROSE,DAN GRAUR,NIR BEN-TAL,et al.Comparison of Site-Specific Rate-Inference Methods for Protein Sequences:Empirical Bayesian Methods are SuperiorMol[J].Biol Evol,2004,21(9):1781-1791.
[20]PENG Kang,PREDRAG RADIVOJAC,SLOBODAN VUCETIC,et al.Length-Dependent Prediction of Protein Intrinsic Disorder[J].BMC Bioinformatics,2006,17(7):208.
[21]KESKIN O,MA B,NUSSINOV R.Hot Regions in Protein-Protein Interactions:the Organization and Contribution of Structurally Conserved Hot Spot Residues[J].J Mol Biol,2005,345(5):1281-1294.
[22]CHO K,KIM D,LEE D.A Feature-Based Approach to Modeling Protein-Protein Interaction Hot Spots[J].Nucleic Acids Res,2009,37(8):2672-2687.
[23]SOL A DEL,FUJIHASHI H,AMOROS D,et al.Residue Centrality,Functionally Important Residues and Active Site Shape:Analysis of Enzyme and Non-Enzyme Families[J].Protein Sci,2006,15(9):2120-2128.
[24]MIHEL J,SIKIC M,TOMIC S,et al.PSAIA-Protein Structure and Interaction Analyzer[J].BMC Struct Biol,2008,21(8):1472-6807.
[25]CHENG J,RANDALL A Z,SWEREDOSKI M J,et al.SCRATCH:a Protein Structure and Structural Feature Prediction Server[J].Nucleic Acids Research,2005,33(1):72-76.
[26]SANNER M F,OLSON A J,SPEHNER J C.Reduced Surface:an Efficient Way to Compute Molecular Surfaces [J].Biopolymers,1996,38(3):305-320.
[27]FU Yan,SUN Ruixiang,YANG Qiang,et al.A Block-Based Support Vector Machine Approach to the Protein Homology Prediction Task in KDD Cup 2004 [J].SIGKDD Explorations,2004,2(6):120-124.
[28]PONOMARENKO J V,BOURNE P E.Antibody-Protein Interactions:Benchmark Dataset and Prediction Tools Evaluation[J].BMC Structural Biology,2007,7(2):64.
[29]MINTSERIS J,WIEHE K,PIERCE B,et al.Protein-Protein Docking Benchmark 2.0:An Update [J].Proteins,2005,60(2):214-216.
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
温州医科大学学报(2019年4期)2019-04-28
中国免疫学杂志(2017年1期)2017-01-17
池州学院学报(2015年3期)2016-01-05
西南医科大学学报(2015年1期)2015-08-22
天津科技大学学报(2015年2期)2015-08-09
畜牧兽医学报(2015年3期)2015-07-05
医学研究杂志(2015年6期)2015-07-01
癌变·畸变·突变(2015年3期)2015-02-27
