
化学修饰的环孢菌素类非免疫抑制性亲环素抑制剂进展
2015-01-07 08:05:28姚贵阳房丽晶潘正银王恒山粟
集成技术 2015年4期
姚贵阳房丽晶潘正银王恒山粟 武
1(中国科学院深圳先进技术研究院 深圳 518055)
2(广西师范大学化学与药学学院 桂林 541000)
化学修饰的环孢菌素类非免疫抑制性亲环素抑制剂进展
姚贵阳1房丽晶1潘正银1王恒山2粟 武1
1(中国科学院深圳先进技术研究院 深圳 518055)
2(广西师范大学化学与药学学院 桂林 541000)
亲环素(Cyps)是一类在自然界分布广泛、具有高度保守性的多功能蛋白家族,也是重要的免疫抑制药物环孢菌素 A(CsA)的蛋白受体,具有肽基脯氨酰顺反异构酶(PPIase)活性。非免疫抑制性亲环素抑制剂是基于CsA 骨架而改造得到的衍生物,这些衍生物能够在抑制亲环素活性的同时降低免疫抑制活性。一些非免疫抑制性亲环素抑制剂(Alisporivir、SCY-635 和 NIM811)已经证明对丙型肝炎传染病治疗有临床疗效。最近的研究发现亲环素蛋白抑制剂还具有治疗多种病毒性感染,炎性症状,以及癌症的潜力。文章综述了非免疫抑制性环孢菌素衍生物在临床和临床前开发的构效关系,并讨论了这些候选药物的药代动力学和化学生物学特性。
亲环素;环孢菌素 A;非免疫抑制;抗病毒;构效关系
1 引 言
环孢菌素 A 是由真菌发酵产生的天然环肽,具有抑制亲环素一类肽基脯氨酰异构酶的活性且一般都有免疫抑制活性[1-3]。对环孢菌素及其类似物的合成和结构修饰衍生出了一些低免疫抑制性但保留抑制亲环素能力的分子[4]。“非免疫抑制性亲环素抑制剂”概念的提出已经为阐明环孢菌素的作用机制和生物特性提供了重要的依据。此外,非免疫抑制性亲环素抑制剂 Alisporivir、SCY-635 和 NIM811 已经在临床上用于治疗丙型肝炎[5]。更值得我们关注的是,很多研究认为这些非免疫抑制性亲环素抑制剂可能对广泛的疾病都有药效。2010-2013 年的报道就明确指出亲环素抑制剂可以用于治疗多种病毒感染、炎性症状、心力衰竭和癌症等疾病[6-9]。
Ito 等[10]在其 2013 年的综述中指出,有一些具有生物活性的环肽又重新应用在医药活性剂上,所以环孢菌素类似物的独特药性和药物特性也得到了重新重视。比如,这些天然类似物清晰的构效关系能够充分说明蛋白与配体亲和力的关系[11],体外亲环素亲和力实验和这些分子在细胞水平的活性研究也提供了寻找提高环肽细胞通透性的方法[12,13]。在本文中,我们讨论了非免疫抑制性环孢菌素的发现及其药代动力学,重点讨论了非免疫抑制性环孢菌素类似物的构效关系。
2 环孢菌素及亲环素的发现
环孢菌素 A(图 1,CsA)最初是由山德士公司在筛选无细胞毒性免疫抑制剂的项目中发现[14,15]。它的开发彻底改变了器官移植的治疗水平,此外,越来越多研究发现环孢菌素也可以应用于银屑病、类风湿关节炎和葡萄膜炎的治疗。深入的机理研究逐渐发现很多环孢菌素免疫抑制作用可以归因于它与细胞亲环素蛋白(Cyps)和钙调磷酸酶(CaN)的两个亚基形成的三元复合物(图 2)[15-17]。因为形成的三元复合物可以保护活化 T 细胞的转录,所以可以阻滞与免疫应答相关的蛋白表达[18,19]。
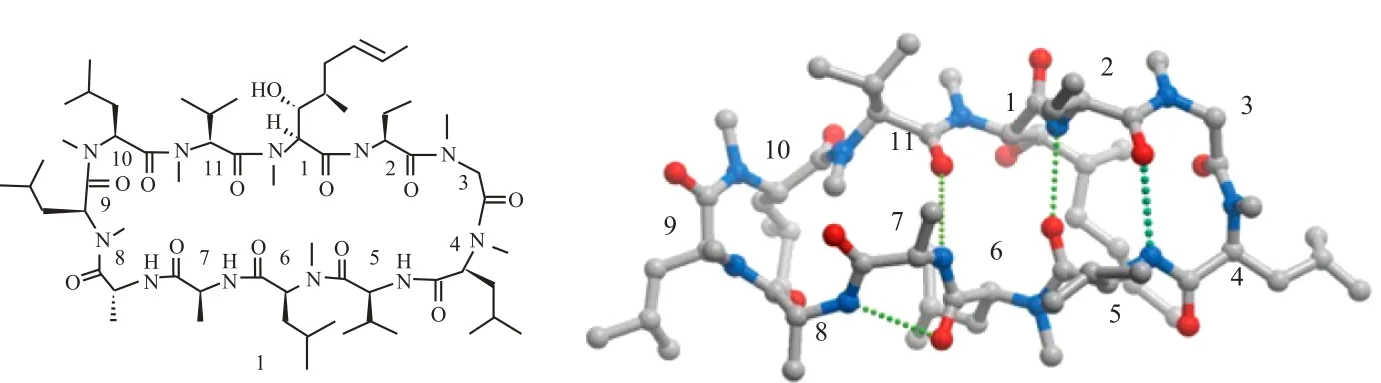
图 1 环孢菌素 A(左,化合物 1)及其骨架结构(右)[15]Fig. 1 Cyclosporin A(left) and its solid state structure(right)[15]
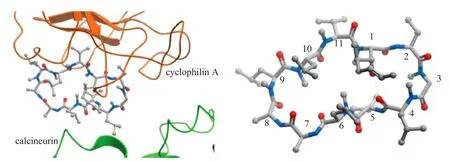
图 2 环孢菌素 A 与亲环素(左)和钙调蛋白(右)的作用示意图(1MF8 和 1CWA))[15]Fig. 2 Left: Complex between cyclophilin A, cyclosporin A, and calcineurin (PDB code 1MF8) Right: Structure of cyclosporin A bound in cyclophilin A conformation (PDB code 1CWA)[15]
2.1 亲环素的发现
亲环素蛋白的发现是基于对天然产物环孢菌素家族的研究。第一个亲环素是由科学家 Fischer等[20]于十九世纪八十年代从猪肾皮质中纯化得到的,在被发现 4 年之后,这个蛋白质被证实与哺乳动物的胸腺细胞[21,22]和人类 T 细胞[23,24]中与环孢菌素 A 结合的蛋白是相同的。目前,Cyps 已经被证明存在于大肠杆菌到智人的各种门类中。比如,酵母的厌氧菌的纤维小体有 8 种亲环素,果蝇有 14 种亲环素,人类细胞至少有 20 种亲环素[25]。其种类目前研究可以分为两大类共 20 种(图 3)[15]。亲环素的原型亲环素 A 几乎是仅有催化结构的低分子量蛋白,其他旁系增加了其他区域可以将分子量提升到 354 kDa,这些区域通过调节蛋白与蛋白的相互作用和细胞内的位置,从而控制这些蛋白质参与许多生物过程[26]。
亲环素是肽基顺反异构酶的一个子集(图3)。这些酶催化顺式和反式脯氨酸的结构互换(图 4)。虽然反式的氨基酸亚氨基的肽键更稳定,但在含脯氨酸(Pro)残基的蛋白质分子中,30% 的肽键是顺式构象。这表明大多数含 Pro 残基的蛋白相互作用需要脯氨酰异构酶。然而,从目前的研究来看,科学家们还不清楚是否所有的生物功能都能被 Cyps 调控。据我们所知自噬蛋白的作用就不需要肽基顺反异构酶活性,因为实验过程中并没有检测到自噬功能涉及的几个肽基脯氨酰顺-反异构酶(PPIL2,PPIL4,PPIL6 和SDCCAG-10)的表达。当 CsA 和 Cyps 结合后,环孢菌素分子结构与游离的 CsA 分子结构相比构象发生了显著的变化[27],分子中原有的氢键全部断裂,新的氢键形成。除此之外,分子中的酰胺键由顺式转化为反式,这样形成的复合物才能与 CaN 结合;而单独的 CsA 或 CyP 则都不能与 CaN 结合。另外,据报道环孢菌素及其类似物与 Cyps 结合能力的强弱并不能体现为免疫抑制的强弱。
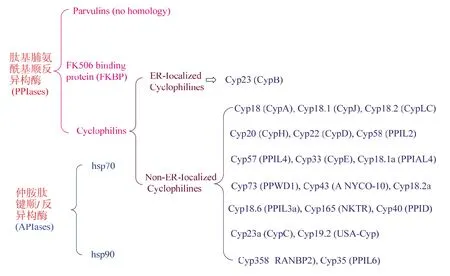
图 3 亲环素蛋白的分类示意图Fig. 3 The class of Cyclophilin protein

图 4 亲环素催化脯氨酸旋转异构体的相互转化示意图Fig. 4 Interconversion of proline rotamers catalyzed by the cyclophilins
2.2 环孢菌素的发现
环孢菌素 A(图 1)是一种分子量约 1200 Da的环状十一肽。各种光谱和结构的研究表明,CsA 及其衍生物的分子结构是非常柔性的。环孢菌素类衍生物与亲环素蛋白的亲和力跟环孢菌素与亲环蛋白结合能密切相关[28-30]。柔性结构也被认为是 CsA 具有显著的细胞通透性和较高的生物利用度的原因[31]。在非极性溶剂和固体(游离的)状态下,CsA 所有的酰胺氢会参与分子内氢键或与酰胺羰基形成氢键作用(图 1,右)。在这种氢键作用下,结构中疏水性侧链和 N-烷基团形成了极性的大环酰胺键疏水层。据推测,环孢菌素A 就是利用这种结构通过细胞膜。CsA 在极性溶剂中结合亲环素时,亲水酰胺基团裸露,肽键处于反式位置。此外,MeBmt 的羟基残基与 P4 位甲基化亮氨酸的羰基形成氢键以保持这些基团的彼此接近从而保持构像的稳定,这是 CsA 能够抑制亲环素的主要原因。
尽管改变 CsA 中氨基酸的种类能影响结合亲环蛋白的活性,但是环孢菌素 A 结合亲环素是直接与 P9、P10、P1 和 P2 基团(见图 1 的编号)的氨基酸相关[32]。环孢菌素 A 和钙调神经磷酸酶之间的亲和作用主要与大环化合物中的 P4、P5 和P6 的氨基酸相关[33]。当 P3 位氮甲基甘氨酸用一个有助于环孢菌素稳定构造代替时,设计非免疫抑制性亲环素抑制剂的一般策略就是修饰 P4-P5-P6位置的氨基酸来减弱与钙调磷酸酶的亲和力。
亲脂化合物环孢菌素 A 在体内广泛分布在组织间隙中。尽管血浆蛋白结合率不是固定值,但是结合率大概为 95%[34]。CsA 和其他亲环素抑制剂的药代动力学分析是非常复杂的[35]。亲环素在血细胞的高度表达(特别是在红细胞)导致亲环素抑制剂在血液/血浆中的配比是剂量依赖的。因此,环孢菌素 A 在红细胞中相对饱和的浓度才能保证完全抑制亲环素。环孢菌素 A 的药理学因为与内源性转运有关联所以更加复杂。在体外和体内,CsA 是有机阴离子转运蛋白的强效抑制剂(OATP1B1 和 OATP1B3)[36,37],也是多药耐药蛋白(MDR1、ABCB1 和 P-gp)、胆盐出口泵的强抑制剂(BSEP)[38]和 MDR2 的抑制剂[39]。通过几个环孢菌素类似物的结构可以推断出该类化合物抑制 MDR1 的构效关系[40]。目前临床上正在研究一种非免疫抑制性环孢素抑制剂伐司朴达(PSC-833)[41],它的潜在药物性质可以抑制肿瘤病人做化疗时产生的副反应[42]。Alisporivir(阿拉泊韦,图 7)[43]是一种与环孢菌素 A 结构高度相似的疏水型非免疫抑制性亲环素抑制剂,也有报道发现其具有抑制 OATP1B1 及 MDR2 活性的效果。这些转运过程一般涉及胆红素的排泄以及胆红素和葡糖苷酸的缀合物排泄到胆汁这些问题。临床研究表明,一些病人服用阿拉泊韦后会得高胆红素血症,所以这类途径的抑制作用可能会引起高胆红素血症。Gallay 等[5]在他的研究中提出大的疏水性环肽往往是 OATP 抑制剂。有趣的是,如下所述,在环孢菌素骨架添加亲水基团可能会减弱对某些转运蛋白的抑制效果。
3 非免疫型环孢菌素
环孢菌素 A 的发现及其潜在的生物活性引起了工业界、学术界以及药物化学界许多科学家的极大兴趣。在山德士公司,苏黎世联邦理工学院教授迪特·泽巴赫和一个由药物化学家组成的团队合作开发了数以百计的环孢菌素类似物的合成方法[44-48]。此外,更多天然存在的环孢菌素类衍生物的发现也使环孢菌素对于抑制细胞免疫应答的构效关系逐渐明朗[49]。亲环素 A 发现之后,酶联免疫吸附测定法发展成为一种可以测定环孢菌素与亲环素结合力的方法[50]。大多数研究报道,多数环孢菌素衍生物的免疫抑制性活性与其结合亲环素 A 的亲和力有关。但是有研究报道在P3 位置修饰大基团虽然保留了 CsA 衍生物与亲环素的亲和力但失去免疫抑制活性性。其中 P3位置修饰的环孢菌素类似物 2 是第一个确定的非免疫抑制性亲环素抑制剂的分子(图 5)[50]。默克公司的科学家也报道了很多化学合成的环孢菌素类似物[51],大部分化合物的亲环素结合力与环孢菌素 A 相当,但是比预期的免疫抑制活性小得多。在他们公布的报告中,化合物 3 就是比较成功的结构改造,改造的方法就是用一个氮甲基丙氨酸取代环孢菌素 A 的氮甲基亮氨酸。更进一步的生物实验表明,化合物 3 具有环孢菌素 A 约50% 的亲环素结合活性,但只有其 0.4% 的免疫抑制活性[52]。更加值得关注的是,有研究报道称亲环素 A 与化合物 3 的复合体对钙调磷酸激酶的亲和力可以忽略不计。1994 年,由山德士公司出版的详细构效关系研究报告就提出了环孢菌素 A 类似物可以在降低钙调磷酸酶的亲和力的同时保留与亲环素结合活性的推论[53]。虽然那时候三元复合体(亲环素/环孢菌素 A/钙调磷酸酶)的晶体结构还没有得到,但是利用核磁共振[54]和环孢菌素 A/亲环素复合体的构造[55]已能够确认 P4-P8 位置是远离亲环素结合分界面的。因此,P4基团的修饰可以寻找并发现环孢菌素A/亲环素复合体和钙调磷酸酶之间的相互作用的构效关系。Wenger 在其研究中报道了环孢素菌 P4 位亮氨酸替代成为缬氨酸(化合物 4,图 6)或者 4'-OH 亮氨酸( 化合物 5,图 6)的化学改造方法,免疫法测定这类衍生物发现这样修饰可以大幅降低免疫抑制活性并保留或改善与亲环素 A 结合能力。从这些结果可以推断出,钙调磷酸酶具有与 P4 位亮氨酸非常紧密结合的氢键作用,而这种假设已经通过共晶实验被证实。
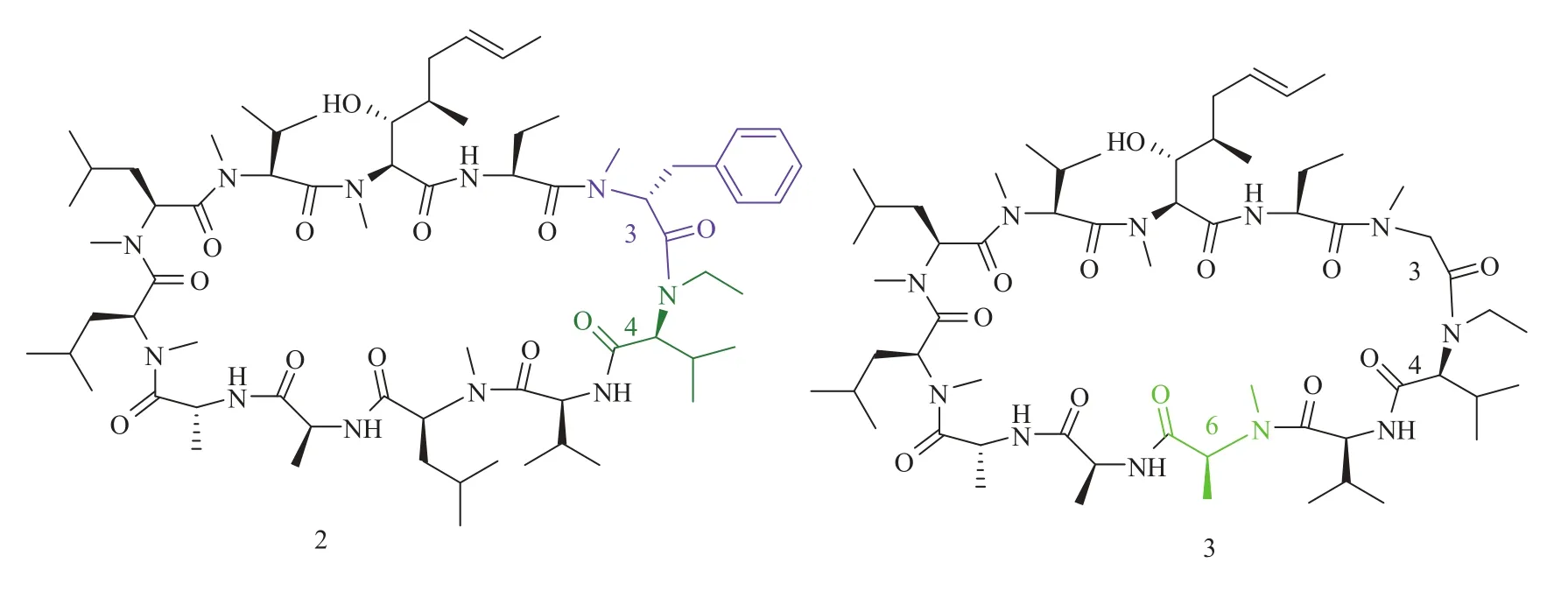
图 5 早期非免疫抑制性亲环素抑制剂(化合物 2 和 3)Fig. 5 Early nonimmunosuppressive cyclophilin inhibitors 2 and 3
3.1 P3 P4 位修饰的环孢菌素
过去的研究已经表明,对 CsA 的 P3 位甘氨酸做适当的修饰[56]或者在 P4 位引入小的疏水性残基[57],可以增加环孢菌素的结合位点从而增强其与亲环素的结合力。这些信息一直指引着药物化学家去寻找比 CsA 更强亲环素 A 亲和力的非免疫抑制性环孢菌素衍生物。其中很多衍生物具有成为抗 HIV 活性的潜在临床药物[58,59]。Lin 的研究就发现代表性类似物 6(图 6)和 NIM-811[60](图7)是 HIV 病毒扩增的抑制剂并且基本上没有免疫抑制活性。有趣的是,在亲环素结合力实验测定中,化合物 6 与亲环素的亲和力较化合物 7 高 4倍,但是它的抗 HIV 活性偏低。这个结果证明用极性残基修饰 CsA 侧链的时候一定要慎重,因为这样修饰可能会影响其细胞通透性[61]。目前对化合物 P3 位的修饰主要还是利用泽巴赫和山德士的方法,也就是使用过量二异丙胺锂(6.0~14.0当量)活化 CsA 的 P3 位置产生烯醇中间体,然后可以与各种亲电基团发生反应(图 8)。
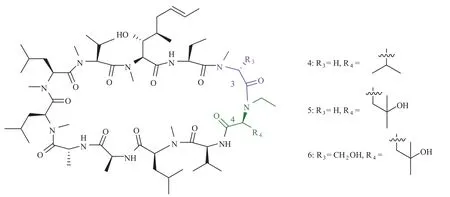
图 6 环孢菌素 P4 位改造的非免疫抑制性亲环素抑制剂(化合物 4、5 和 6)Fig. 6 P4-modified nonimmunosuppressive cyclophilin inhibitors 4,5 and 6
从这些最初的研究中获得的先导化合物来看,大多数衍生物具有比 CsA 更高的结合亲环素 A 的活性。这些抑制剂已经作为丙肝的潜在临床药物进行研究,除此之外也使用于许多其他疾病的细胞和动物模型中。其中化合物 NIM-811的发现似乎早于上述构效关系的详细研究中,因为它最初是作为副产物从弯颈霉属病菌的菌株发酵产生的环孢菌素类似物中分离的。菌株的基因操作是大规模地获得化合物 NIM-811 的主要原因,其方法是直接分离有氮甲基异亮氨酸的微生物发酵液。化合物 NIM-811 的抗 HIV 活性是因为它可以和干扰亲环素 A 与 HIV-p24 核心蛋白结合。随着发现环孢菌素 A 在丙肝病毒(Hepatitis C Virus,HCV)复制中的特异作用,化合物 NIM-811 的发展被专注于丙肝肝炎的治疗上[62,63]。值得我们关注的是,尽管 HCV 具有高度遗传变异性,但是化合物 NIM-811 仍然可以有效阻止其耐药性的快速发展。这些研究说明非免疫抑制性亲环蛋白抑制剂具有限制高基因屏障的丙肝病毒耐药性的产生。在临床一期治疗研究中,化合物NIM-811 在 HCV 感染病人中进行了抗病毒活性的研究[64]。虽然每日两次进量且剂量高达 600 毫克,但是在 14 天之后病人没有明显抗病毒治疗的副反应。
化合物 NIM-811 在许多疾病的动物模型中被当作亲环素抑制剂使用。结果发现大多数疾病的发生过程与亲环素功能有关。在四氯化碳处理生成肝纤维化的动物模型中,化合物 7的使用可以降低肝坏死的程度[65]。这种活性归因于化合物NIM-811 对亲环素 B 和亲环素 F 的抑制活性。这类化合物在小鼠诱导胆汁淤积模型和减少肝损伤的肝切除术中可以减少胆汁淤积。这种结果是因为化合物 NIM-811 具有通过抑制肝细胞线粒体转换孔的形成来抑制线粒体凋亡的能力[66]。其他研究还认为化合物 NIM-811 在体外的实验是可以起到保护神经的作用[67,68]。而在小鼠哮喘模型中,化合物 NIM-811 被发现具有修复白细胞活化的作用。此外抗炎活性的研究发现细胞外的环孢菌素在白细胞迁移中扮演了绝对重要的角色[69]。
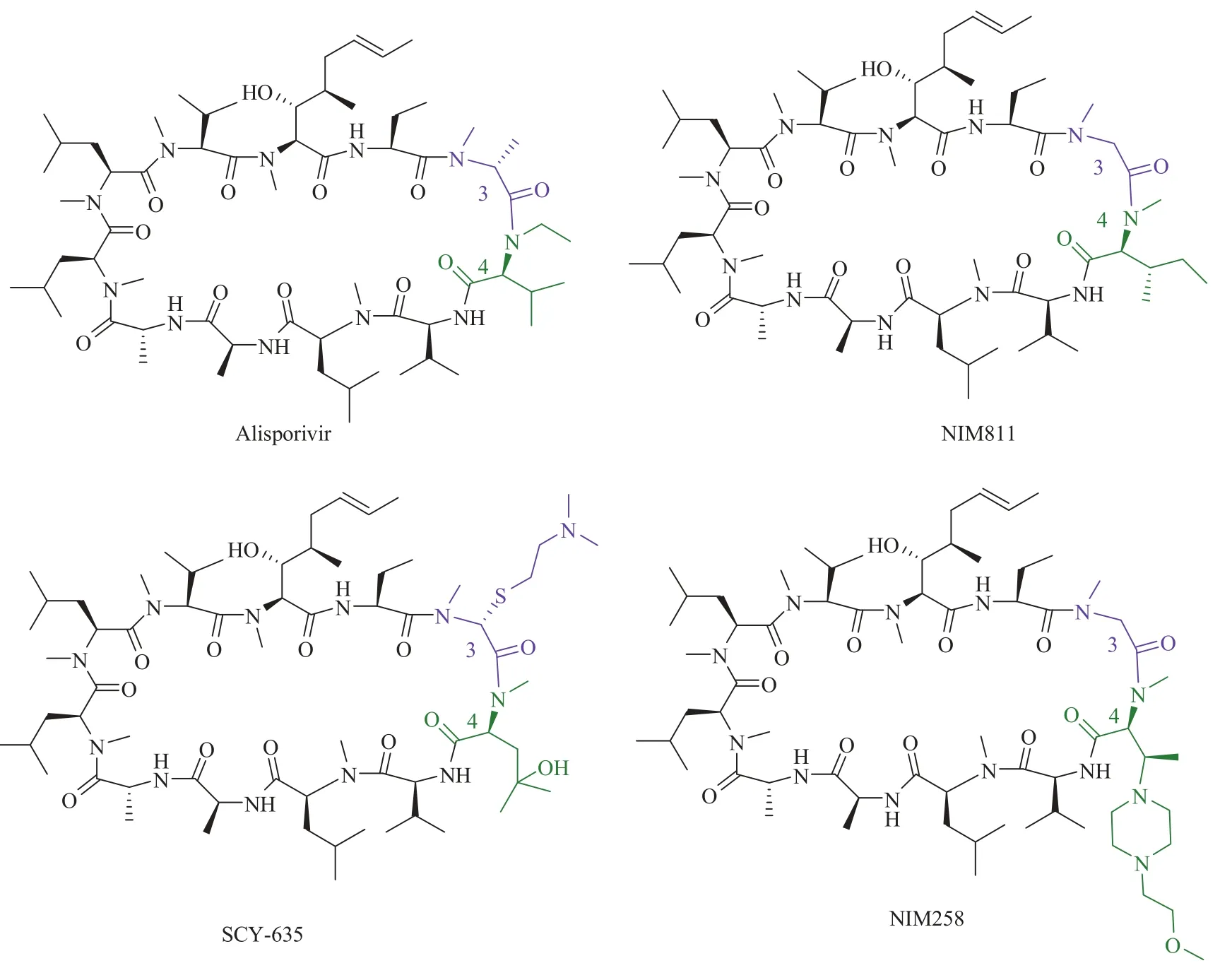
图 7 化合物 Alisporivir、NIM811、SCY-635 和 NIM258 的结构Fig. 7 Structure of Alisporivir, NIM811, SCY-635 and NIM258
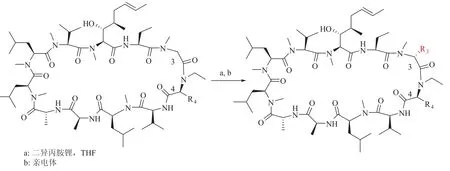
图 8 P3 烯醇化合物的生成与修饰Fig. 8 P3 enolate generation and modification
Wenger 和他的团队研发了一种可以使环孢菌素 A 在 P3 和 P4 两个位置同时改造的合成方法(图 9)[70]。该方法首先在碱性环境下将大环环肽从 P3 和 P4 之间开环,然后通过埃德蒙降解法将 P4 的残基脱掉。然后在这个新的线性肽末端通过酰胺耦合引入新的想改造的 P3 和 P4。通过巧妙的切割和重排反应,原先 P3 位置的甲基化甘氨酸被脱掉。最后,新的 P3-P4 的修饰线性肽在偶联剂和较低浓度的条件下进行环化从而得到一系列 CsA 的衍生物。Debiopharm SA 公司和洛桑大学的科学家采用这种技术,细致研究了这个位置的构效关系,提出一个新的非免疫抑制性亲环素抑制剂 Debio-025(Alisporivir[71]图 7)。在山德士公司早期的研究中,化合物 Alisporivir 中 P4位的缬氨酸基团和 P3 位 D-丙氨酸残基可以减弱衍生物与钙调磷酸酶的亲和力。此外,在 P4 位还做了一个氮乙基的改造,对于包括 CsA 在内的一些亲环素抑制剂来讲,这个 P4 N-Me 基团也是CYP3A4 介导代谢的主要位点[72]。用 N-Et 代替N-Me 可以提高 CsA 衍生物的生物利用度。

图 9 环孢菌素在 P3 和 P4 位改造的方法Fig. 9 Method for modification of P3 and P4 of cyclosporins
对环孢菌素衍生物抗 HCV 活性的研究发现:Alisporivir 抑制 HCV 的活性大约比 CsA高 5~10 倍[73];此外,在细胞水平的抗病毒分析中发现其比化合物 NIM811 活性还高。化合物 Alisporivir 的抗病毒活性之所以比早期的一代亲环素抑制剂活性高,可能是因为 P3 位的改造可以提高 Alisporivir 与亲环素的结合力。用 Alisporivir 作用后丙肝病毒的耐药性发展缓慢[74],而且当该化合物与直接作用的抗病毒剂结合使用时,肝脏细胞会迅速清晰地复制。Alisporivir 的改造最初是用来研究治疗艾滋病毒感染。对感染艾滋病毒的患者进行 10 天给药研究显示,每日一次给药都可以降低病人血浆中的HIV-1RNA 浓度。随后在 HIV-HCV 合并感染患者的研究中,科学家们加大了剂量并对患者的变化进行了研究。结果证明在较高剂量下药物释放并没有按照线性方式增加[75]。
环孢菌素衍生物 SCY-635(图 7)是一种非免疫抑制性亲环素抑制剂,目前已经进入治疗 HCV 感染的临床二期。该化合物最初是由Aventis 公司在研究 HIV 治疗的亲环素抑制剂时发现的[76]。通过 Sebekia benihana 菌株的酶催化作用得到了 P4 位是 4-羟基-N-甲基亮氨酸的 CsA 衍生物。然后选择性的烷硫化在 P3 位引入巯基化合物。在这个系列衍生物中,科学家发现化合物 SCY-635 的 P3 取代基从-SCH3延伸到-SCH2CH2N(CH3)2基团时,化合物的抗 HIV活性会增加,同时体外免疫抑制活性会降低[77]。化合物 SCY-635 的临床前研究发现该化合物抑制亲环素 A 的活性只是稍微比化合物 CsA高。此外,对其抗 HCV 活性的研究发现其体外抑制 HCV 复制是随时间变化而变化的,但直到给药 72 小时后才可以完全抑制 HCV 的复制。相比许多其他亲环素抑制剂来说,化合物SCY-635 似乎具有较弱的抑制转运蛋白活性。尽管在中等浓度(15 μmol/L)下就可以观察到的 P-糖蛋白外排活性的完全抑制,但在浓度大约是 150 μmol/L 时其抑制 MRP2 介导的转运作用依然十分弱[78]。
Enanta 制药公司 Owens 等[79]开发了一个从 CsA 中衍生出的非免疫抑制性亲环素抑制剂EDP-546(其结构没有报道)。2014 年年初,该公司已经启动了该分子临床 I 期的研究,虽然化合物 EDP-546 的结构尚未发布,但从专利申请发现其结构应该高度类似于 NIM258 的结构(图 9)。在临床前药代动力学研究中,化合物 10(图 10)有较低的血液清除率并能高效地分配到肝脏的各个部位。在水性缓冲溶液中,可以观察到其具有良好的溶解性,特别是在 pH 低于 7 的缓冲液中。另外,化合物 10(图 10)对胆红素、药物转运蛋白 MRP2 和 OATP1B1 的抑制作用比化合物8(图 10)弱得多[80]。S&T Global 公司 Su 等[81-84]也制备了一系列的 P3 和 P4 改造的 CsA 衍生物,多数的化合物是以 P4 位 4-OH-Me 亮氨酸环孢菌素为起点去改造。大部分衍生物在细胞水平的测定中显示有抗 HCV 的活性。艾尔健公司也报道了一系列 P3 位置修饰的 CsA 衍生物[85]。结合安万特公司和山德士集团的最初报告可以发现,当CsA 的 P4 位亮氨酸基团保留的时候,某些 P3 位替换可能会降低与钙调磷酸酶的亲和力。
3.2 P1 位改造的环孢菌素类衍生物
MeBmt 氨基酸对环孢菌素 A 与亲环素的结合力的大小是非常重要的,原因在于这个基团从亲环素结合界面延伸到非常接近钙调磷酸酶的结合区域(图 2)。较早的研究工作表明,MeBmt位的改造在保留亲环素的结合活性的同时往往没有办法保留 CsA的免疫抑制活性[86]。比较突出的例子就是默克集团推出的化合物[CH(OH) CH(CH3)-CH2SCH3]P1-CsA,该衍生物只有 CsA 10% 的免疫抑制活性,但是亲环素结合活性比CsA 高 78%。在这之后,Aurinia 公司报道一个MeBmt 基团增长了碳链且 P3 位置原来的甲基化甘氨酸改为 D-N-Me-Ala 的 CsA 衍生物 7(图 10)[87]。研究发现,该分子是非免疫型亲环素抑制剂,且抑制亲环素 A 的活性比 CsA 高 13 倍。更加不可思议的是,其免疫抑制活性只有 CsA 的 3%。在2013 年 7 月,Aurinia 公司已经在抗丙肝病毒活性的充分评估范围内证实该化合物是一个先导候选药物。
一般来讲,测定环孢菌素衍生物的免疫抑制活性是在分离的 T 细胞中。在这种情况下,环孢菌素的免疫抑制活性主要是与亲环素 A-环孢菌素复合体抑制钙调磷酸酶的活性有关。然而,如上所述,环孢菌素也会参与其他免疫反应过程,比如先天免疫和其他的炎症。为了探索亲环素抑制剂是否具有通过抑制细胞外的亲环素抑制白细胞运输的潜能,德国科学家 Cordelia Schiene-Fischer 开发了一系列渗透细胞膜较差的苯并咪唑类环孢菌素衍生物[61]。其中,代表化合物 8(图10)在体外脯氨酸异构酶结合测定中被证实与亲环素 A 结合力和 CsA一样,而一个使用荧光标记的的亲环素衍生物与细胞内的亲环素结合分析显示化合物 8 的细胞膜通透性比 CsA 低 50 倍左右。但是令人惊喜的是,尽管化合物 8 主要是限制在细胞外间隙的,但是化合物 8 依然能抑制亲环素 A 进而抑制白细胞的迁移。在小鼠模型中发现这个抑制剂还可以减少白细胞的活化。
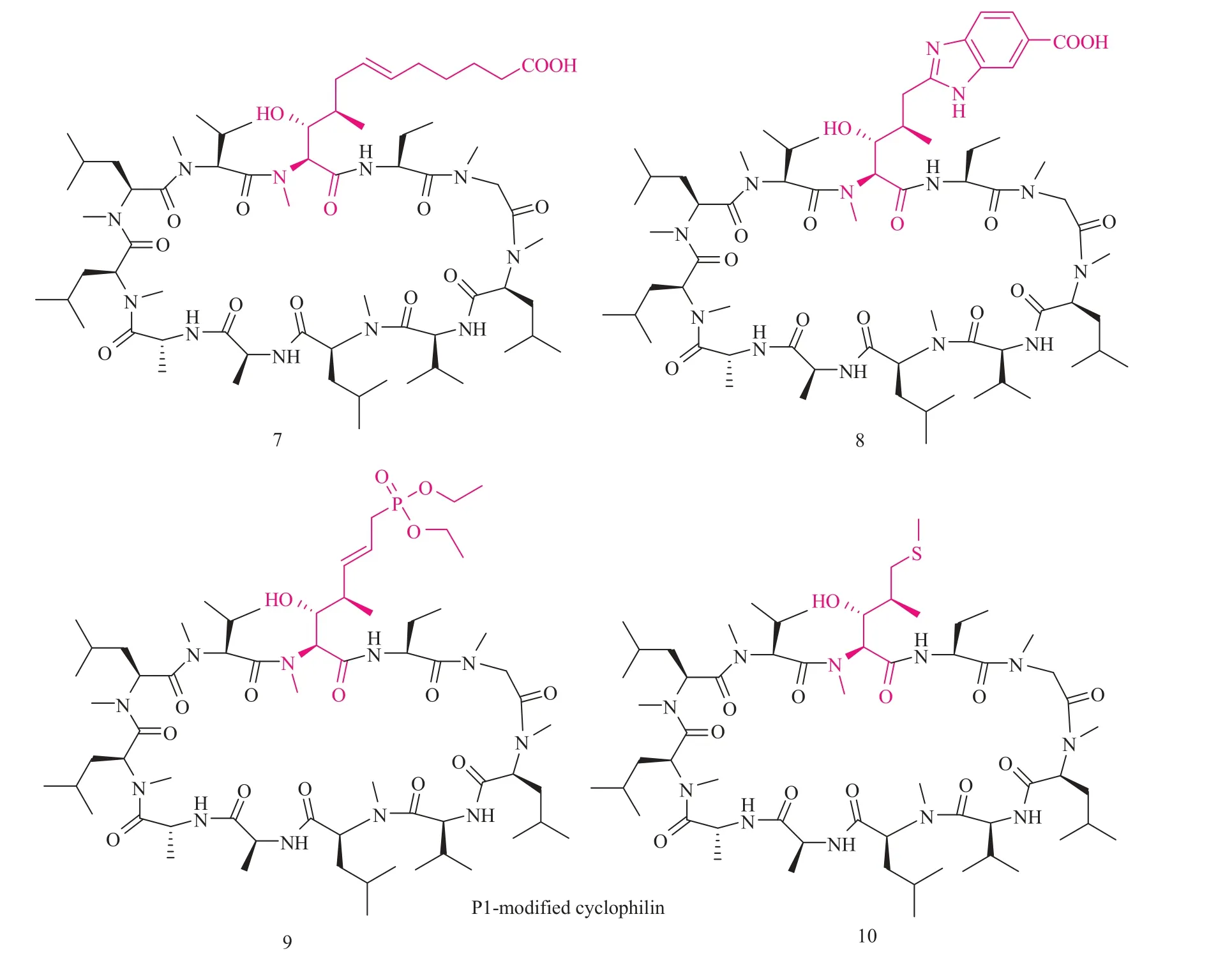
图 10 化合物 7、8、9 和 10 的结构Fig. 10 Structure of compound 7, 8, 9 and 10
3.3 其他位置改造的环孢菌素类衍生物
虽然 Scynexis 公司探索 P5 位烷基化影响CsA 的免疫抑制活性和抗病毒活性的专利数据不详。 但在上世纪末山德士公司的科学家已经证明这个位置的游离 NH 基的烷基化可以影响环孢菌素衍生物的构像和免疫抑制活性[88,89]。化合物11(图 11)就是通过二甲基烯丙基溴的烷基化来合成,这个 CsA 衍生物被报道对亲环素 A 和 D有较好的亲和力(<60 nmol/L)。因为像 P3 氨基酸、P8 氨基酸已经被定位在钙调磷酸酶(P4-P7)结合域的边缘,所以 Scynexis 公司使用类似于 Eberle公司探索 P8 位置修饰的环孢素所介绍的方法(图12)研究了这个位置的构效关系[90,91]。他们的研究结果建议可以通过在这个位置接入碱性的基团来改造亲环素 A 结合活性、免疫抑制活性和抗病毒活性。研究发现代表性的化合物 12(图 11)的抗HCV 活性比 CsA 高且免疫抑制活性也低于 CsA 。
4 影响环孢菌素与亲环素和钙调磷酸激酶亲和力因素
环孢菌素 A 结构的 P1、P2、P9、P10 和 P11氨基酸是环孢菌素 A 与 CyP 的结合区,所以修饰这些位置的氨基酸往往可以提高环孢菌素衍生物对亲环素的结合力。P4-P7 位的氨基酸是与 CaN作用的效应区,CsA 和 CyP 的结合是产生抑制CaN 活性的基础,修饰上述位置往往会影响环孢菌素与亲环素的构像,但是更大程度上会改变其结合钙调磷酸激酶的活性。
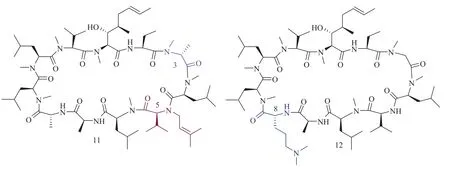
图 11 化合物 11 和 12 的结构Fig. 11 Structure of compound 11 and 12
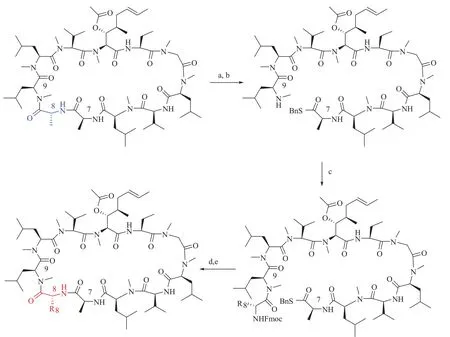
图 12 环孢菌素在 P8 位改造的方法Fig. 12 Production of P8-modified cyclosporins
在环孢菌素 A 与 CyP 的结合区中对亲和作用有重要贡献的是 P1 位 MeBmt 氨基酸,MeBmt氨基酸结构与 CyP 的最适底物结构相似,形成的过渡态能量最低最稳定,这对 CsA 和 CyP 的高亲和力是很重要的。一般来说 MeBmt 的改造往往会降低 CsA 衍生物对亲环素的亲和力。例如删掉或改变 MeBmt 中甲基立体构型,甚至是增加一个甲基都会降低其亲和力。同样如果 MeBmt的羟基去掉或者被保护都会降低其与亲环素亲和力。最近研究发现,当利用生物电子等排体 S 取代双键位置,其与亲环素的亲和力会增加 78%。具体的结构改造结果可见图 13。
如上文所述,对 CsA 的结构改造热点是寻找高效低毒非免疫抑制的亲环素抑制剂,其关键就是减弱甚至去除钙调磷酸激酶的亲和力。因为 P4 位置甲基化亮氨酸与钙调磷酸激酶的 Trp352和 Phe356有氢键作用,所以最有效的办法可能就是改造 P4位置的氨基酸。研究报道将亮氨酸换为异亮氨酸的时候,这个衍生物(NIM-811)就失去了免疫抑制活性,但是仍然保留着与 CsA 相似的亲环素结合活性。具体的 P4-P7 位置的构效关系总结在图 13 中。
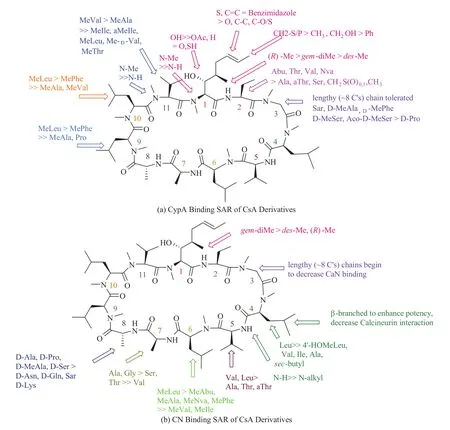
图 13 环孢菌素与亲环素和钙调磷酸激酶的构效关系Fig. 13 CypA and CaN binding SAR of CsA derivatives
5 结论与展望
自从在 30 年前发现第一个免疫抑制性环孢菌素类似物,药物化学家对有临床效应的亲环素抑制剂的安全鉴定有了巨大的进步,环孢菌素的结构修饰帮助科学家进一步发现环孢菌素未知的生物功能。已经有了几个此类的分子进入到用于病毒感染治疗的临床试验。为了更进一步了解亲环素抑制剂的结构多样性,更多的非免疫抑制环孢菌素衍生物需要开发。
在这过程中发现的更好的化合物会被安排进入治疗丙型肝炎感染或心肌再灌注损伤治疗的临床试验中。据我们所知,所有研究发现的亲环素抑制剂至少都是亲环素 A、B 和 F 的有效抑制剂,一个选择性抑制亲环素的分子急需开发出来。非环孢菌素类亲环素抑制剂的开发可能会是寻找选择性亲环素抑制剂的趋势。目前许多亲环素脯氨酰异构酶活性的小分子拮抗剂已被报道,我们热切期待这些分子的出现引领新一代生化工具和临床药物的发展[92]。
2010 年,西班牙药物化学家 Muñoz 等[93]研究了环孢菌素 A 的广谱抗肿瘤活性,发现其对人脑胶质瘤、神经母细胞瘤、视网膜母细胞瘤和胰腺、喉、胃及结肠癌细胞都具有较强的细胞毒性,并发现这些作用主要介导 NK-1 受体而诱导肿瘤细胞凋亡。2012 年,韩国科学家 Jeon 等[94]研究了环孢菌素 A 对前列腺癌的抑制作用。结果发现其具有较强的抑制作用并阻滞细胞周期在 G1 期。深入的凋亡机制发现 CsA 是通过 CaMKII 介导而激活 AMPK 从而抑制前列腺癌细胞 mTORC1 信号杀死前列腺癌细胞。这些工作为 CsA 往抗肿瘤方向的发展开辟了先河。
[1] Davis TL, Walker JR, Campagna-Slater V, et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases [J]. PLoS Biology, 2010, 8(7): e1000439.
[2] Borel JF, Di Padova F, Mason J, et al. Pharmacology of cyclosporine(Sandimmyne) [J]. Pharmacological Reviews, 1989, 41: 239-242.
[3] Sanglier JJ, Quesniaux V, Hofmann H, et al. Sanglifehrins A, B, C and D, novel cyclophilinbinding compounds isolated from Streptomyces sp. A92-308110. I. Taxonomy, fermentation, isolation and biological activity [J]. The Journal of Antibiotics, 1999,52(5): 466-473.
[4] Peel M, Scribner A. Optimization of cyclophilin inhibitors for use in antiviral therapy [M] // Desai MC, Meanwell NA.Successful Strategies for the Discovery of Antiviral Drugs. Cambridge: Royal Society of Chemistry, 2013: 384-418.
[5] Baugh J, Gallay P. Cyclophilin involvement in the replication of hepatitis C virus and other viruses [J]. Biological Chemistry, 2012, 393(7): 579-587.
[6] Frausto SD, Lee E, Tang H. Cyclophilins as modulators of viral replication [J]. Viruses, 2013, 5(7): 1684-1701.
[7] Yurchenko V, Constant S, Eisenmesser E, et al. Cyclophilin-CD147 interactions: a new target for anti-inflammatory therapeutics [J]. Clinical and Experimetal Immunology, 2010, 160(3): 305-317.
[8] Gill RS, Bigam DL, Cheung PY. The role of cyclosporine in the treatment of myocardial reperfusion injury [J]. Shock, 2012, 37(4): 341-347.
[9] Lee JH, Kim SS. Current implications of cyclophilins in human cancers [J]. Journal of Experimental and Clinical Cancer Research, 2010, 29(1): 97.
[10] Ito K, Passioura T, Suga H. Technologies for the synthesis of mRNA-encoding libraries and discovery of bioactive natural product-inspired nontraditional macrocyclic peptides [J]. Molecules, 2013, 18(3): 3502-3528.
[11] Schärfer C, Schulz-Gasch T, Ehrlich HC, et al. Torsion angle preferences in druglike chemical space: a comprehensive guide [J]. Journal of Medicinal Chemistry, 2013, 56(5): 2016-2028.
[12] Bockus AT, McEwen CM, Lokey RS. Form and function in cyclic peptide natural products: a pharmacokinetic perspective [J]. Current Topics in Medicinal Chemistry, 2013, 13(7): 821-836.
[13] Alex A, Millan DS, Perez M, et al. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space [J]. Medicinal ChemistryCommunication, 2011, 2(7): 669-674.
[14] Heusler K, Pletscher A. The controversial early history of cyclosporin [J]. Swiss Medical Weekly, 2001, 131(21-22): 299-302.
[15] Sweeney ZK, Fu JP, Wiedmann B. From chemical tools to clinical medicines: nonimmunosuppressive cyclophilin inhibitors derived from the cyclosporin and sanglifehrin scaffolds [J]. Journal of Medicinal Chemistry, 2014, 57(17): 7145-7159.
[16] Columbani PM, Ross A, Hess AD. Cyclosporin a binding to calmodulin: a possible site of action on T lymphocytes [J]. Science, 1985, 228(469): 337-339.
[17] Schreiber SL. Immunophilin sensitive protein phosphatase action in cell signaling pathways minireview [J]. Cell, 1992, 70(3): 365-368.
[18] Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506 [J]. Immunology Today, 1992, 13(4): 136-142.
[19] Peel M, Scribner A. Cyclophilin inhibitors as antiviral agents [J]. Bioorganic & Medicinal Chemistry Letters, 2013, 23(16): 4485-4492.
[20] Fischer G, Bang H, Mech C. Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides [J]. Biomedica Biochimica Acta, 1984, 43(10): 1101-1111.
[21] Handschumacher RE, Harding MW, Rice J, et al. Cyclophilin: a specific cytosolic binding protein for cyclosporin A [J]. Science, 1984, 226(4674): 544-547.
[22] Harding MW, Handschumacher RE, Speicher DW. Isolation and amino acid sequence of cyclophilin [J]. The Journal of Biological Chemistry, 1986, 261(18): 8547-8555.
[23] Fischer G, Wittmann-Liebold B, Lang K, et al. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins [J]. Nature, 1989, 337(6206): 476-478.
[24] Takahashi N, Hayano T, Suzuki M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin [J]. Nature, 1989, 337(6206): 473-475.
[25] Galat A. Function-dependent clustering of orthologues and paralogues of cyclophilins [J]. Proteins, 2004, 56(4): 808-820.
[26] Galat A, Bua J. Molecular aspects of cyclophilins mediating therapeutic actions of their ligands [J]. Celluler and Molecular Life Sciences, 2010, 67(20): 3467-3488.
[27] Fu JP, Tjandra M, Becker C, et al. Potent nonimmunosuppressive cyclophilin inhibitors with improved pharmaceutical properties and decreased transporter inhibition [J]. Journal of Medicinal Chemistry, 2014, 57(20): 8503-8516.
[28] Wenger RM, France J, Bovermann G, et al. The 3D structure of a cyclosporin analogue in water is nearly identical to the cyclophilin-bound cyclosporin conformation [J]. FEBS Letters, 1994, 340(3): 255-259.
[29] Taylor P, Husi H, Kontopidis G, et al. Structures of cyclophilin-ligand complexes [J]. Progress in Biophysics and Molecular Biology, 1997, 67(2-3): 155-181.
[30] Rosen MK, Belshaw PJ, Alberg DG, et al. The conformation of cyclosporin a bound to cyclophilin is altered(once again) following binding to calcineurin: an analysis of receptor-ligandreceptor interactions [J]. Bioorganic & Medicinal Chemistry Letters, 1992, 2(7): 747-753.
[31] O’Donohue MF, Burgess AW, Walkinshaw MD, et al. Modeling conformational changes in cyclosporin A [J]. Protein Science, 1995, 4(10): 2191-2202.
[32] Mikol V, Kallen J, Pflügl G, et al. X-ray structure of a monomeric cyclophilin A-cyclosporin A crystal complex at 2.1 A resolution [J]. Journal of Molecular Biology, 1993, 234(4): 1119-1130.
[33] Jin L, Harrison SC. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin [J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99(21): 13522-13526.
[34] Akhlaghi F, Trull AK. Distribution of cyclosporin in organ transplant recipients [J]. Clinical Pharmacokinetics, 2002, 41(9): 615-637.
[35] Awni WM, Sawchuk RJ. The pharmacokinetics of cyclosporine II. Blood plasma distribution and binding studies [J]. Drug Metabolism and Disposition, 1985, 13(2): 133-138.
[36] Amundsen R, Christensen H, Zabihyan B, et al. Cyclosporine A, but not tacrolimus, shows relevant inhibition of organic anion-transporting protein 1B1-mediated transport of atorvastatin [J]. Drug Metabolism and Disposition, 2010, 38(9): 1499-1504.
[37] Shitara Y, Nagamatsu Y, Wada S, et al. Long-lastinginhibition of the transporter-mediated hepatic uptake of sulfobromophthalein by cyclosporin a in rats [J]. Drug Metabolism Disposition, 2009, 37(6): 1172-1178.
[38] Ogimura E, Sekine S, Horie T. Bile salt export pump inhibitors are associated with bile aciddependent drug-induced toxicity in sandwichcultured hepatocytes [J]. Biochemical and Biophysical Research Communications, 2011, 416(3-4): 313-317.
[39] El-Sheikh AA, Greupink R, Wortelboer HM, et al. Interaction of immunosuppressive drugs with human organic anion transporter(OAT) 1 and OAT3, and multidrug resistance-associated protein (MRP) 2 and MRP4 [J]. Translation Research, 2013, 162(6): 398-409.
[40] Loor F, Tiberghien F, Wenandy T, et al. Cyclosporins: structure-activity relationships for the inhibition of the human MDR1 P-glycoprotein ABC transporter [J]. Journal of Medicinal Chemistry, 2002, 45(21): 4598-4612.
[41] Kolitz JE, George SL, Marcucci G, et al. P-glycoprotein inhibition using valspodar(PSC-833) does not improve outcomes for patients younger than age 60 years with newly diagnosed acute myeloid leukemia: Cancer and Leukemia Group B study 19808 [J]. Blood, 2010, 116(9): 1413-1421.
[42] Friedenberg WR, Rue M, Blood EA, et al. Phase III study of PSC-833(valspodar)in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95): a trial of the Eastern Cooperative Oncology Group [J]. Cancer, 2006, 106(4): 830-838.
[43] Kovacs SJ, Ke J, Dabovic K, et al. Therapeutic dose effects of the cyclophilin inhibitor alisporivir (ALV) on circulating bilirubin concentrations in healthy subjects [J]. Gastroenterology, 2012, 142(5 Supplement 1): S964.
[44] Wenger RM. Synthesis of cyclosporine. Total syntheses of “cyclosporin A” and “cyclosporin H”, two fungal metabolites isolated from the species Tolypocadium inflatum GAMS [J]. Helvetica Chimica Acta, 1984, 67(2): 502-525.
[45] Wenger RM. Synthesis of cyclosporin and its analogs: relation between structure and immunosuppressive activity [J]. Angewandte Chemie International Edition in English, 1985, 24(2): 77-85.
[46] Wenger RM, Payne TG, Schreier MH. Cyclosporine: chemistry, structure-activity relationships and mode of action [C] // Metabolic Control in Diabetes Mellitus Beta Adrenoceptor Blocking Drugs NMR Analysis of Cancer Cells Immunoassay in the Clinical Laboratory Cyclosporine, Progress in Clinical Biochemistry and Medicine, 1986, 3: 157-191.
[47] Seebach D, Bossler H, Gruendler H, et al. C-alkylation of peptides through polylithiated and lithium chloride solvated derivatives containing sarcosine lithium enolate units [J]. Helvetica Chimica Acta, 1991, 74(1): 197-224.
[48] Ko SY, Wenger RM. Solid-phase total synthesis of cyclosporine analogues [J]. Helvetica Chimica Acta, 1997, 80(3): 695-705.
[49] Von Traber R, Hofmann H, Loosli HR, et al. Neue cyclosporine aus tolypocladium inflatum die cyclosporine K-Z [J]. Helvetica Chimica Acta, 1987, 70(1): 13-36.
[50] Quesniaux VF, Schreier MH, Wenger RM, et al. Cyclophilin binds to the region of cyclosporine involved in its immunosuppressive activity [J]. European Journal of Immunology, 1987, 17(9): 1359-1365.
[51] Sigal NH, Dumont F, Durette P, et al. Is cyclophilin involved in the immunosuppressive and nephrotoxic mechanism of action of cyclosporin A? [J]. The Journal of Experimental Medicine, 1991, 173(3): 619-628.
[52] Liu J, Albers MW, Wandless TJ, et al. Inhibition of T cell signaling by immunophilin-ligand complexes correlates with loss of calcineurin phosphatase activity [J]. Biochemistry, 1992, 31(16): 3896-3901.
[53] Papageorgiou C, Borer X, French RR. Calcineurin has a very tight-binding pocket for the side chain of residue 4 of cyclosporin [J]. Bioorganic & Medicinal Chemistry Letters, 1994, 4(2): 267-272.
[54] Thériault Y, Logan TM, Meadows R, et al. Solution structure of the cyclosporin A/cyclophilin complex by NMR [J]. Nature, 1993, 361(6407): 88-91.
[55] Pflügl G, Kallen J, Schirmer T, et al. X-ray structure of a decameric cyclophilin-cyclosporin crystal complex [J]. Nature, 1993, 361(6407): 91-94.
[56] Zeder-Lutz G, Van Regenmortel MH, WengerR, et al. Interaction of cyclosporin A and two cyclosporin analogs with cyclophilin: relationship between structure and binding [J]. Journal of Chromatography B: Biomedical Sciences and Applications, 1994, 662(2): 301-306.
[57] Papageorgiou C, Florineth A, Mikol V. Improved binding affinity for cyclophilin a by a cyclosporin derivative singly modified at its effector domain [J]. Journal of Medicinal Chemistry, 1994, 37(22): 3674-3676.
[58] Papageorgiou C, Sanglier J, Traber R. Anti HIV-1 activity of a hydrophilic cyclosporin derivative with improved binding affinity to cyclophilin A [J]. Bioorganic & Medicinal Chemistry Letters, 1996, 6(1):23-26.
[59] Billich A, Hammerschmid F, Peichl P, et al. Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus(HIV) type 1: interference with HIV protein-cyclophilin A interactions [J]. Journal of Virology, 1995, 69(4): 2451-2461.
[60] Lin K. Discovery of cyclophilin inhibitor NIM811 as a novel therapeutic agent for HCV [M] // Kazmierski WM. Antiviral Drugs: From Basic Discovery Through Clinical Trials. Hoboken: Wiley, 2011: 317-329.
[61] Malesevic M, Gutknech D, Prell E, et al. Antiinflammatory effects of extracellular cyclosporins are exclusively mediated by CD147[J].Journal of Medicinal Chemistry, 2013, 56(18): 7302-7311.
[62] Teraoka S, Mishiro S, Ebihara K, et al. Effect of cyclosporine on proliferation of non-A, non-B hepatitis virus [J]. Transplantation Proceedings, 1988, 20(3 Suppl 3): 868-876.
[63] Goto K, Watashi K, Murata T, et al. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811 [J]. Biochemical and Biophysical Research Communications, 2006, 343(3): 879-884.
[64] Lawitz E, Godofsky E, Rouzier R, et al. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy [J]. Antiviral Research, 2011, 89(3): 238-245.
[65] Wang H, Zhang Y, Wang T, et al. N-methyl-4-isoleucine cyclosporine attenuates CCl-induced liver fibrosis in rats by interacting with cyclophilin B and D [J]. Jouranl of Gastroenterology and Hepatology, 2011, 26(3): 558-567.
[66] Rehman H, Ramshesh VK, Theruvath TP, et al. NIM811(N-methyl-4-isoleucine cyclosporine), a mitochondrial permeability transition inhibitor, attenuates cholestatic liver injury but not fibrosis in mice [J]. The Journal of Pharmacology and Experimental Therapeutics, 2008, 327(3): 699-706.
[67] Hokari M, Kuroda S, Iwasaki Y. Pretreatment with the ciclosporin derivative NIM811 reduces delayed neuronal death in the hippocampus after transient forebrain ischaemia [J]. The Journal Pharmacy and Pharmacology, 2010, 62(4): 485-490.
[68] Korde AS, Pettigrew LC, Craddock SD, et al. Protective effects of NIM811 in transient focal cerebral ischemia suggest involvement of the mitochondrial permeability transition [J]. Journal of Neurotrauma, 2007, 24(5): 895-908.
[69] Yurchenko V, Constant S, Bukrinsky M. Dealing with the family: CD147 interactions with cyclophilins [J]. Immunology, 2006, 117(3): 301-309.
[70] Hubler F, Rückle T, Patiny L, et al. Synthetic routes to NEtXaa4-cyclosporin A derivatives as potential anti-HIV I drugs [J].Tetrahedron Letters, 2000, 41(37): 7193-7196.
[71] Wenger RM, Mutter M, Ruckle T. Novel cyclosporin with improved activity profile:PCT/ IB1999/001232 [P/OL]. 2000-01-13 [2015-03-15]. https://patentscope.wipo.int/search/en/detail. jsf?docId=WO2000001715.
[72] Radeke HH, Christians U, Sewing KF, et al. The syntergistic immunosuppressive potential of cyclosporin etabolite combinations [J]. International Journal of Immunopharmacology, 1992, 14(4): 595-604.
[73] Paeshuyse J, Kaul A, De Clercq E, et al. The nonimmunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro [J]. Hepatology, 2006, 43(4): 761-770.
[74] Coelmont L, Hanoulle X, Chatterji U, et al. DEB025(Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A [J]. PLoS One, 2010, 5(10): e13687.
[75] Nicolas V. PK-PD modelling and simulation from a biotech company perspective [C] // ThePharmacokinetics UK 2010 Meeting, 2010.
[76] Evers M, Barrière JC, Bashiardes G, et al. Synthesis of non-immunosuppressive cyclophilin-binding cyclosporin A derivatives as potential anti-HIV-1 drugs [J]. Bioorganic & Medicinal Chemistry Letters, 2003, 13(24): 4415-4419.
[77] Seebach D, Beck AK, Bossler HG, et al. Modification of cyclosporin A(CS): generation of an enolate at the sarcosine residue and reactions with electrophiles [J]. Helvetica Chimica Acta, 1993, 76(4): 1564-1590.
[78] Wring S, Wille K, Rewerts C, et al. In vitro models for assessing the relative risk of hyperbilirubinemia associated with cyclophilin inhibitor therapy [J]. Journal of Hepatology, 2010, 52(Supplement 1): S263.
[79] Owens CM, Brasher BB, Polemeropoulos A, et al. EP-CyP546 is a potent non-immunosuppressive cyclophilin inhibitor with an excellent preclinical profile [J]. Hepatology, 2011, 54: 541A-542A
[80] Jiang LJ, Liu S, Phan TU, et al. EDP-546, a potent and novel cyclophilin inhibitor with favorable preclinical pharmacokinetic and safety profiles [J]. Hepatology, 2012, 56: 1076A-1076A.
[81] Su Z, Long ZY, Huang ZN, et al. Novel cyclosporin derivatives for the treatment and prevention of viral infections: PCT/US2012/051572 [P/OL]. 2013-02-28 [2015-03-15]. https://patentscope.wipo.int/ search/en/detail.jsf?docId=WO2013028615.
[82] Su Z, Long ZY, Huang ZN, et al. Novel cyclosporin derivatives for the treatment of a viral infection: PCT/US2011/047571 [P/OL]. 2012-02-16 [2015-03-15]. https://patentscope.wipo.int/search/en/ detail.jsf?docId=WO2012021796.
[83] Su Z, Long ZY, Huang ZN, et al. Novel cyclosporin derivatives for the treatment and prevention of a viral infection: PCT/ US2011/063295 [P/OL]. 2012-06-07 [2015-03-15]. https://patentscope.wipo.int/search/en/detail. jsf?docId=WO2012075494.
[84] Su Z, Huang ZN, Long ZY, et al. Novel cyclosporin derivatives for the treatment and prevention of a viral infection: PCT/ US2010/044362 [P/OL]. 2012-01-19 [2015-03-15]. https://patentscope.wipo.int/search/en/detail. jsf?docId=WO2012009715.
[85] Carling WR, Scowen DA, Garst ME, et al. Cyclosporin analogs: PCT/US2011/055788 [P/OL]. 2012-04-19 [2015-03-15]. https:// patentscope.wipo.int/search/en/detail. jsf?docId=WO2012051193.
[86] Búa J, Ruiz AM, Potenza M, et al. In vitro antiparasitic activity of cyclosporin A analogs on trypanosoma cruzi [J]. Bioorganic & Medicinal Chemistry Letters, 2004, 14(18): 4633-4637.
[87] Hegmans A, Fenske BW, Trepanier DJ, et al. Cyclosporine analogue molecules modified at amino acid 1 and 3: PCT/CA2011/050773 [P/OL]. 2012-06-21 [2015-03-15]. https://patentscope.wipo.int/ search/en/detail.jsf?docId=WO2012079172.
[88] Li K, Peel MR. Novel cyclic peptides: PCT/ GB2010/052057 [P/OL]. 2011-06-16 [2015-03-15]. https://patentscope.wipo.int/search/en/detail. jsf?docId=WO2011070364.
[89] Papageorgiou C, Kallen J, France J, et al. Conformational control of cyclosporin through substitution of the N-5 position. A new class of cyclosporin antagonists [J]. Bioorganic & Medicinal Chemistry, 1997, 5(1): 187-192.
[90] Eberle MK, Hiestand P, Jutzi-Eme AM, et al. Preparation and in vitro activities of ethers of [D-serine]8-cyclosporin [J]. Journal of Medicinal Chemistry, 1995, 38(11): 1853-1864.
[91] Scribner A, Houck D, Huang Z, et al. Synthesis and biological evaluation of[D-lysine]8cyclosporin A analogs as potential anti-HCV agents [J]. Bioorgganic & Medicinal Chemistry Letters, 2010, 20(22): 6542-6546.
[92] Guichou JF, Colliandr L, Ahemed-Belkacem H, et al. New inhibitors of cyclophilins and uses thereof: PCT/EP2010/070359 [P/OL]. 2011-06-03 [2015-03-15]. https://patentscope.wipo.int/search/en/ detail.jsf?docId=WO2011076784.
[93] Muñoz M, Rosso M, González A, et al. The broadspectrum antitumor action of cyclosporin A is due to its tachykinin receptor antagonist pharmacological profile [J]. Peptides, 2010, 31(9): 1643-1648.
[94] Lee CR, Chun JN, Kim SY, et al. Cyclosporin A suppresses prostate cancer cell growth through CaMKKβ /AMPK-mediated inhibition of mTORC1 signaling [J]. Biochemical Pharmacology, 2012, 84(4): 425-431.
Nonimmunosuppressive Cyclophilin Inhibitors Derived from the Cyclosporin Scaffolds
YAO Guiyang1FANG Lijing1PAN Zhengyin1Wang Hengshan2SU Wu1
1(Shenzhen Institutes of Advanced Technology,Chinese Academy of Sciences,Shenzhen518055,China)
2(School of Chemistry & Pharmaceutical Sciences of Guangxi Normal University,Guilin541004,China)
Cyclophilins (Cyps) are crucial to protein folding because that the ubiquitous proteins affect the cis-trans isomerization of Pro amide bonds. The immunosuppressive natural product cyclosporine A inhibits the enzymatic activity of the cyclophilins. Chemical modification of cyclosporine scaffolds has produced many analogues that inhibit cyclophilins in vitro but have reduced immunosuppressive properties. Three nonimmunosuppressive cyclophilin inhibitors (alisporivir, SCY-635, and NIM811) have demonstrated clinical efficacy for the treatment of hepatitis C infection. Recent publications suggest that cyclophilin inhibitors may have utility for the treatment of diverse viral infections, inflammatory indications, and cancer. In this review, we document the structure-activity relationships of the nonimmunosuppressive cyclosporins in clinical and preclinical development. Aspects of the pharmacokinetic behavior and chemical biology of these drug candidates are also described.
cyclophilins; cyclosporine A; nonimmunosuppressive; antiviral; structure-activity relationships
R 914.5
A
2015-04-21
:2015-05-10
深圳市引进海外高层次人才“孔雀计划”(KQCX20130628112914285);国家自然科学基金(21432003)
姚贵阳,博士研究生,研究方向为多肽药物的合成及开发;房丽晶,博士,助理研究员,研究方向为多肽药物的合成及开发;潘正银,硕士,多肽药物的合成及开发;王恒山,博士,教授,研究方向为手性药物的开发研究;粟武(通讯作者),博士,研究员,研究方向新药研发以及磁性纳米材料应用于医学成像和药物靶向运输, E-mail:wu.su@siat.ac.cn。
猜你喜欢
现代畜牧科技(2021年7期)2021-07-28 06:41:00
当代水产(2021年3期)2021-07-20 07:20:44
兽医导刊(2019年1期)2019-02-21 01:13:54
新闻传播(2018年11期)2018-08-29 08:15:30
新闻传播(2018年13期)2018-08-29 01:06:52
新闻传播(2016年9期)2016-09-26 12:20:34
癌症进展(2016年12期)2016-03-20 13:16:17
中国药理学与毒理学杂志(2015年3期)2015-12-16 09:11:40
新闻传播(2015年7期)2015-07-18 11:09:57
现代检验医学杂志(2015年1期)2015-02-06 01:59:04
