
不同工艺条件下CO甲烷化催化剂的稳定性
2015-01-07 03:37魏雪梅唐浩东刘化章
化学反应工程与工艺 2015年5期
魏雪梅,唐浩东,李 鑫,刘化章
浙江工业大学工业催化研究所,浙江 杭州 310014
不同工艺条件下CO甲烷化催化剂的稳定性
魏雪梅,唐浩东,李 鑫,刘化章
浙江工业大学工业催化研究所,浙江 杭州 310014
为开发性能较优的合成气甲烷化催化剂,采用固相混合法制备了20%Ni/Al2O3催化剂,通过H2程序升温还原(H2-TPR)和X射线衍射(XRD)表征发现,简单固相混合法制备的催化剂具有较好的分散性和还原性能。考察了压力、温度、空速和进料比(H2与CO的体积比)等不同操作条件下该CO甲烷化催化剂的稳定性,结果表明:在CO甲烷化反应体系中随着温度、压力和进料比的增大,催化剂稳定性增强;空速增大,催化剂稳定性降低。采用XRD和H2程序升温表面反应(H2-TPSR)技术对催化剂失活原因进行了探讨,发现催化剂由于表面沉积无定形胶质碳(Cβ)而失活,升高温度和压力会使催化剂表面活性碳物种(Cα)向更稳定的蠕虫状碳(Cv)和石墨型碳(Cc)沉积,从而催化剂稳定性增强。
一氧化碳 甲烷化催化剂 催化稳定性
煤制天然气的核心技术是煤气化技术和甲烷化技术,故开发国内自主产权且具有较优性能的甲烷化催化剂是煤制天然气技术的核心问题之一。Ni基催化剂是目前国内外最常用的甲烷化催化剂[1-3],但在甲烷化反应中,不同反应条件下催化剂呈现的活性[4]、催化剂失活的速率[5,6]、催化剂的稳定性[7]有很大不同。托普索公司在上个世纪七八十年代就发现催化剂存在β-失活现象[8,9],近年Nguyen等[3]报道了反应后催化剂失活原因分析,发现随着温度从330 ℃升高到370 ℃,催化剂表面胶质碳减少,失活现象减轻。Barrientos等[10]在研究不同载体甲烷化催化剂时也发现在310 ℃催化剂活性不稳定现象,并认为适当改变工艺条件有利于减缓失活。研究表明,积炭可能会使镍基催化剂活性下降,但积炭类型和位置对催化剂性能影响更大[11]。
Bartholomew等[12]对甲烷化过程中催化剂表面的积炭类型和形貌进行了总结(见表1),认为主要有以下几种:吸附型碳原子(Cα),无定形胶质碳(Cβ),蠕虫状碳(Cv),镍的碳化物(Cγ),石墨型碳(Cc)。

表1 Ni基催化剂上CO分解积炭形成不同碳物种形貌及反应活性[12]Table 1 Forms and reactivity of carbon species formed by CO decomposition on nickel
不同类型的碳形成温度不同,活性不同,与H2反应时的温度也不同。Cα形成温度通常在325 ℃以下,是活泼炭,能与H2在较低温度下反应,也会随着反应时间和温度的增加会进一步转化为Cβ或Cv等。当Cα的生成速率过大,在镍表面来不及溶解扩散或加氢时,就会在镍表面沉积生成Cβ,为一种包覆型[10]的胶质碳,覆盖催化剂活性中心,使催化剂失活(β-失活),这是催化剂低温失活的主要原因。选择合适的评价条件不仅可以筛选出性能优良的催化剂,还可以考察工艺条件对催化剂稳定性的影响,在实际工业化过程中就可以尽量避开催化剂易失活的操作条件,从而延长催化剂寿命。因此,本研究将详细考察工艺条件对甲烷化催化剂活性、选择性和稳定性能的影响,为催化剂工业化操作提供参考依据。
1 实验部分
1.1 催化剂制备和表征
取一定量氧化铝,置于马弗炉中800 ℃下焙烧4 h,冷却至室温,得到所需氧化铝载体。称取9.909 g的Ni(NO3)2·6H2O,研细后和载体混合均匀,置于马弗炉中350 ℃焙烧4 h,冷却至室温,制得20%NiO/Al2O3催化剂,筛分后取60~100 目待用。
H2程序升温还原-质谱联用(H2-TPR-MS)自搭反应装置测定催化剂的还原性能。称取0.10 g催化剂,在200 ℃用Ar吹扫60 min后降至室温,通入5%H2-95%Ar混合气(30 mL/min),从室温以10 ℃/min升至850 ℃,在线质谱(英HIDEN QIC-20)同步检测耗氢量。
采用H2程序升温表面反应-质谱联用(H2-TPSR-MS)自搭反应装置分析反应后催化剂表面积炭类型。称取50 mg催化剂,在150 ℃用A(r30 mL/min)吹扫60 min后降至室温,通入H(230 mL/min),以10 ℃/min从室温升至850 ℃,在线质谱(英HIDEN QIC-20)同步检测产生甲烷量。
X射线衍射(XRD)测定在瑞士Themo ARL公司SCINTAG X’TRA型X射线衍射仪上进行。Cu Kα辐射源,管电流40 mA,管电压40 kV,扫描速度4.0 (°)/min,扫描2θ为20~80 °。
1.2 催化剂性能评价
催化剂评价在合成气制甲烷反应在四管固定床反应装置上进行(反应管内径12 mm)。取催化剂60~100目,用同等粒度石英砂按体积比1:9稀释,并置于反应器等温区。催化剂在2.0 MPa,450 ℃下用H2还原4 h。活性测试条件为:H2与CO的体积比为3.0,压力2.0 MPa,温度350 ℃,体积空速(GHSV)为24 000 h-1,催化剂体积(Vcatal)为0.5 mL,在该评价条件下采用单一变量法考察工艺条件对催化剂稳定性影响。当反应温度为500 ℃时,不锈钢反应管内加玻璃衬管以防止不锈钢反应管对评价产生影响。尾气产物在美国Agilent公司的7890A气相色谱仪进行,检测柱为13X柱、P-Q柱、P-N柱和5A柱等,H2、CO、CO2、CH4采用热导(TCD)在线检测分析,产物中的气相烃采用氢火焰(FID)在线检测分析,外标法定量。CO转化率(XCO)、H2转化率(XH2)、气相产物CH4、CO2、C2+的选择性(Si)及单位体积合成气CH4收率(YCH4)的计算公式如下:

式中F表示气体体积流量,mL/min;yi表示组分i的体积分数。
2 结果与讨论
2.1 催化剂表征
图1是催化剂还原前后的XRD图。从图1可以看出还原前催化剂在2θ为37.5,45.5,67.2 °处出现了γ-Al2O3的衍射峰,在37.5,43.2,62.9,75.3,79.5 °处出现了NiO的衍射峰,说明还原前催化剂由NiO和γ-Al2O3两种物相组成,没有发现固熔体。对2θ为43.2 °的NiO衍射峰进行拟合,得NiO的晶粒度约21.5 nm,说明固相混合法制备的催化剂氧化镍的分散较好[13,14],这可能是因为硝酸镍在氧化铝上自发分散的原因。催化剂在450 ℃还原后NiO衍射峰消失,只有金属Ni和γ-Al2O3的衍射峰,说明Ni/Al2O3催化剂在450 ℃基本可以还原完全。对2θ为44.5 °的Ni的衍射峰进行拟合,得到Ni的平均粒径约21.6 nm,说明具有较好的分散度。
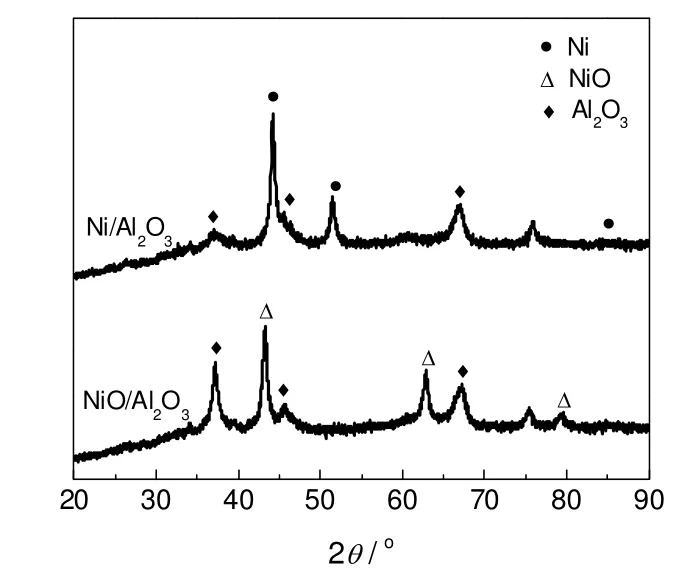
图1 还原前后Ni/Al2O3催化剂XRD图谱Fig.1 XRD patterns of Ni/Al2O3catalyst before and after reduction

图2 Ni/Al2O3催化剂H2-TPR图谱Fig.2 H2-TPR profile of Ni/Al2O3catalyst
图2是氧化态Ni/Al2O3催化剂H2-TPR图。一般[15,16]认为Ni/Al2O3催化剂中,NiO存在三类不同的晶型,即α-NiO、β-NiO和γ-NiO。从图中可以看出,还原过程出现四个还原峰,分别反映了这三类不同晶型NiO的还原:第一个峰顶温度245 ℃,属于表面的游离氧化镍即α-NiO的还原峰,温度为200~300 ℃左右;第二个峰顶温度339 ℃,还原温度在300~350 ℃左右,是与载体相互作用较弱的氧化镍(β1-NiO)的还原;第三个峰顶温度400 ℃,还原温度在350~475 ℃左右,是与载体表面相互作用较强的氧化镍(β2-NiO)的还原峰;第四个峰顶温度为550 ℃,还原温度在475 ℃以上,可归属为富铝的氧化镍即镍铝尖晶石(γ-NiO)的还原峰。γ-NiO为高温还原峰,该物种对催化剂活性贡献不大[17]。因此,在本研究催化剂性能评价实验中,选用的还原条件为2.0 MPa,450 ℃,H2流量60 mL/min。
2.2 不同工艺条件下的催化剂性能
2.2.1 温度的影响
图3是不同温度下Ni/Al2O3催化剂的甲烷化性能考察结果。从图3中的初活性数据来看,350 ℃和500 ℃下催化剂在起初CO转化率都约100%,但温度从350 ℃升高到500 ℃,催化剂的H2转化率从98%降到了81%,CH4选择性从96%降低到了84%,CH4收率从23%降低到了20%。这是因为甲烷化是强放热反应,升高温度在热力学上是不利的,故其活性和选择性随温度升高而降低,这与热力学计算[18,19]趋势相符。从图3(a)还可以看出,当温度为350 ℃,催化剂逐渐失活,而当温度升高到500 ℃时,催化剂活性虽较350 ℃稍有下降,但具有很好的稳定性。

图3 不同温度下的催化剂稳定性Fig.3 The performance of Ni/Al2O3under different temperature
图4是不同温度反应前后催化剂XRD图,可以看出,与反应前催化剂对比,500 ℃反应后未失活的催化剂在2θ为26 °时出现了明显的石墨碳物种的衍射峰,而350 ℃反应后失活的催化剂在2θ为26 °时只有微弱的碳物种衍射峰,由此可知石墨碳物种并不是引起催化剂失活的原因。在衍射峰2θ为44.5 °处,对镍的晶粒大小进行拟合得,350,500 ℃反应后镍的晶粒度分别是24.5,23.7 nm,与反应前的21.6 nm相比,催化剂晶粒度均稍增大,但不同温度反应后的催化剂镍的晶粒度并无明显差别,结合不同温度下催化剂的稳定性不同可知,在实验条件下,催化剂Ni的晶粒大小的变化不是导致350 ℃反应后催化剂失活的原因,即350 ℃反应时催化剂失活并非因烧结而引起的。

图4 不同温度反应后催化剂XRD图谱Fig.4 XRD profiles of the used catalysts

图5 不同温度反应后催化剂H2-TPSRFig.5 H2-TPSR profiles of the used catalysts
催化剂H2-TPSR技术可用来研究催化剂表面的积炭现象。图5是不同温度反应后催化剂的H2-TPSR图,曲线与横坐标间的面积可表示催化剂表面的积炭量,与H2反应的峰温可表示表面碳物种的反应活性,从而可区分不同类型积炭。从图5可以看出当温度为350 ℃时,主要存在两种类型积炭:峰温在200~350 ℃左右的积炭,为表面活性碳(Cα),峰温在500 ℃左右积炭,为包覆型胶质碳(Cβ),会覆盖催化剂表面活性位,使催化剂失活。当温度为500 ℃时,出现大量低温加氢峰(<350 ℃),说明催化剂表面存在大量活性更高的表面活性碳,易使催化剂失活的峰温在500 ℃左右积炭(Cβ)明显较少,而且在600 ℃左右出现了归属于须状碳(Cv)的耗氢峰,700 ℃左右出现了石墨碳的耗氢峰,与图4中石墨碳(Cc)物种的衍射峰相符。说明催化剂在高温时反应时表面积炭进一步向更稳定形式的Cv和Cc发生了转变,这可能是催化剂在500 ℃依然保持较高稳定性的原因。
根据图5中反应后催化剂H2-TPSR曲线可拟合得到峰面积数据,除以在线评价时间即得到350 ℃和500 ℃的相对积炭速率为5.65E-8、1.67E-8,表明500 ℃时催化剂表面积炭速率小于350 ℃。以CO为碳源时催化剂表面积炭是以下四个反应之间的平衡[20]:① CO在Ni表面解离吸附形成Cα,② Cα加氢生成CH4,③ Cα在Ni表面沉积生成Cβ,④ Cα向体相扩散生成Cv及Cc,其中②③④三个反应是竞争反应。350 ℃反应时甲烷化体系中Cα的生成速率较快,表面Cα在镍表面来不及溶解扩散或加氢时,就会在镍表面沉积生成Cβ,为一种包覆型[3,12]的胶质碳,覆盖催化剂活性中心,使催化剂失活(β-失活);而500 ℃反应时,Cα在催化剂镍金属表面沉积速率较慢,这可能由以下两个原因造成:1)表面活性碳(Cα)随温度增加加氢速率增加,使Cα生成包覆型胶质碳(Cβ)选择性下降;2)在高温下Cα向镍体相中的溶解扩散速率增加,大于Cα在镍表面的沉积速率,从而提高向体相沉积,生成具有须状、纤维状结构的积炭(Cv)的选择性,避免生成Cβ,故催化剂不易失活。且催化剂表面积炭的类型比积炭量对催化剂活性影响更大[21]。
2.2.2 压力的影响
图6是350 ℃、不同压力下催化剂性能随时间的变化。从图6中可以看出,不同压力下催化剂甲烷化反应初活性差别不大,但稳定性有很大差别。由图6(a)可知,当压力从0.1 MPa增加到5 MPa,CO转化率保持在100%的时间依次为6,16,132,306 h;而当压力为5.0 MPa时,CO转化率一直保持在100%,并未下降。从图6(b)和(f)来看,当压力为0.1,2.0,3.0 MPa时,催化剂H2转化率和甲烷收率从反应开始一直在下降,且随着压力减小,下降速度加快,说明在0.1~3.0 MPa下催化剂活性不稳定,在逐渐失活,且随着压力减小,催化剂失活加快。当压力提高至5.0 MPa时,催化剂活性一直保持很稳定。而CH4的选择性随着压力减小先降低后升高,CO2和C2+的选择性先升高后降低,可能原因是随着催化剂逐渐失活,生成甲烷的活性位减少,相应生成CO2和C2+,故甲烷选择性先降低,CO2和C2+选择性先升高;而当催化剂已经基本失活后,生成CO2和C2+的活性位也很少,故此时造成CH4的选择性升高,CO2和C2+的选择性降低。从图6可以看出,压力5 MPa时,催化剂CO和H2转化率一直稳定在100%和92%左右,CH4的选择性稳定在92%左右,CO2选择性仅有1%左右,较其它压力下小,C2+的选择性约8%,即在5 MPa下反应时,催化剂活性和选择性一直很稳定。综上所述,在本实验的高空速(GHSV为24 000 h-1)条件下,提高压力有利于提高催化剂的稳定性、CH4选择性和CH4时空收率;并有利于降低CO2的选择性。
图7和图8是不同压力反应后催化剂的XRD和H2-TPSR结果。从图7中可以看出,0.1,2.0 MPa反应后催化剂物相无明显变化,3.0 MPa反应后的催化剂在2θ为26 °处出现了微弱的碳物种衍射峰,5.0 MPa反应后催化剂在2θ为26 °处出现了明显的石墨衍射峰,但随着压力增加,催化剂稳定性增强,说明石墨碳物种并不会引起催化剂失活。而对不同压力反应后的催化剂在衍射峰2θ为44.5 °处,对镍的晶粒大小进行拟合,得0.1,2.0,3.0 MPa反应后失活的催化剂镍的晶粒度分别是22.8,24.5,23.8 nm,而5.0 MPa反应后未失活催化剂镍的晶粒度为25.7 nm。这说明,在评价条件下催化剂失活与其晶粒度大小并无必然联系,故不同压力反应后催化剂失活并非因烧结而引起的。

图7 不同压力反应后催化剂XRD图谱Fig.7 XRD profiles of the used catalysts under different pressures

图8 不同压力反应后催化剂H2-TPSRFig.8 H2-TPSR profiles of the used catalysts under different pressure
而由图8可知,反应后催化剂表面积炭量及其表面碳与H2的反应活性与压力有关。随着压力增加,催化剂表面积炭量增加。当压力为0.1,2.0 MPa时,催化剂表面主要存在两种积炭类型,分别归属[12]于Cα和Cβ:峰温在250 ℃左右的Cα碳在反应条件下可以加氢生成甲烷,为表面活性碳;峰温在450 ℃左右Cβ碳,在反应条件下不能和氢气发生反应,是一种胶质碳,这类胶质碳易覆盖催化剂表面活性位,使催化剂失活。
故当压力为0.1,2.0 MPa时,尽管积炭总量不大,但催化剂活性持续下降,稳定时间较短。当压力增大至3.0,5.0 MPa时,催化剂表面积炭大部分进一步转化为蠕虫状碳(Cv)和石墨型碳(Cc)。当压力升高,催化剂稳定性增强的可能原因是催化剂表面H2和CO存在竞争[22,23]吸附,压力升高,对H2的吸附增加更快,催化剂表面碳加氢速率增快,从而避免了催化表面碳的沉积;另一方面是随着压力升高催化剂表面碳向体相沉积加快,生成更稳定形式的Cv和Cc,这两类积炭对催化剂活性无影响,故使得催化剂稳定性增强。根据Gao等[20,21]热力学计算结果可知,随着压力升高,C的选择性降低,因而在热力学上升高压力对催化剂的稳定性也是有利的。
由以上分析可知,虽然在低压下催化剂甲烷化反应具有很高的活性,但催化剂寿命欠佳。故适当升高压力可以明显增加催化剂稳定性。
2.2.3 原料气进料比的影响
图9是在350 ℃、3.0 MPa的条件下,不同进料比(H2与CO的体积比)对催化剂性能影响评价结果。从图9(a)、(b)和(f)来看,随着进料比从1增加到4,催化剂稳定性增强:当进料比为1时,催化剂在反应6 h时CO和H2的转化率仅为10%左右,可能原因是催化剂失活速率过快,在开始测样时就已经基本失活;当进料比为2时,在30 h内CO转化率迅速下降到15%左右;进料比为3时,CO转化率维持在100%,且稳定性较好,但从60 h开始,CO和H2转化率开始下降,并逐渐失活;进料比增加到4时,在评价时间(110h)内催化剂CO转化率一直稳定在100%,但从图9(b)和(f)可以看出,与稳定期进料比为3的数据相比,H2转化率低了20%左右,甲烷收率也低了约5%。

图9 不同进料比时的Ni/Al2O3催化剂性能Fig.9 The performance of Ni/Al2O3catalysts under different volume ratios of H2to CO
图9的结果表明,增加进料比,催化剂稳定性会增加,但当进料比增加到4时,催化剂H2和CH4收率都会大幅降低。Ponec[22]认为CO和H2在催化剂表面形成竞争吸附,且CO比H2吸附更强,故当进料比较低时,CO在催化剂表面大量解离吸附形成积炭,使催化剂失活,而升高进料比有助于增加H2在催化剂表面的吸附量,使表面活性碳加氢生成甲烷。故考虑甲烷收率和催化剂的寿命可控制氢气和一氧化碳气体的进料比略大于3,从而增强催化剂稳定性,提高甲烷收率。
为了探讨不同进料比反应后催化剂失活原因,对反应后的催化剂进行了XRD分析,结果见图10。可见,进料比为1时的催化剂在2θ为26 °处出现了碳物种的衍射峰,随着进料比增加其碳物种衍射峰强度减弱,当进料比为4时,反应后催化剂表面无碳物种的峰。在衍射峰2θ为44.5 °处对镍的晶粒大小进行拟合,得进料比为1、2、3的反应后催化剂的镍晶粒度分别是25.3,24.4,23.8 nm,而进料比为4时反应后未失活催化剂镍的晶粒度为23.3 nm。可以看出,在评价条件下,催化剂失活并非因Ni晶粒度变化即烧结而引起的。

图10 不同进料比反应后催化剂XRD图谱Fig.10 XRD profiles of the used catalysts under different volume ratios of H2to CO

图11 不同进料比反应后催化剂H2-TPSR图谱Fig.11 H2-TPSR profiles of the used catalysts under different volume ratios of H2to CO
图11是不同进料比反应后催化剂H2-TPSR图,结合表2的拟合数据可知:进料比为1时的催化剂表面只存在非活性的胶质碳(Cβ),积炭量为7.3E-7,Cβ覆盖在催化剂活性中心,使催化剂失活;进料比为2时的催化剂表面存在非活性的胶质碳(Cβ)和须状碳(Cv),积炭量分别是1.3E-6、5.0E-7,且积炭总量比进料比为1时要大;当进料比为3时,催化剂表面存在积炭类型较多,且各种类型的碳量都是最大,积炭总量也最大,可能是表面形成大量Cα,且在进料比为3时并无过量的氢气使表面Cα加氢生成甲烷,故Cα向催化剂体相溶解扩散,在350 ℃时其溶解扩散速率小于其生成速率,故在催化剂表面沉积形成Cβ,覆盖催化剂金属活性位,使催化剂逐渐失活;当进料比为4时,催化剂表面存在大量活性碳(Cα)和须状碳(Cv),Cα为甲烷化反应活性中间体,当反应体系中H2分压增大时,Cα加氢速率加快,表面存在Cα的量就降低,且随着Cα向催化剂Ni金属体相扩散,逐渐形成Cv,虽然Cv的量在逐渐增大,但其量2.0E-7依然很小,且对催化剂的活性并没有影响,故催化剂最稳定,没有任何失活的迹象。催化剂表面积炭是由Cα的生成、加氢、表面沉积、体相溶解扩散四者共同决定[24],当进料时氢碳比增加时,催化剂稳定性增强,当氢碳比大于3时,催化剂具有较好的稳定性。

表2 不同进料比反应后催化剂表面碳归属及H2-TPSR峰面积Table 2 The fitting result of H2-TPSR curves of the used catalysts under different volume ratios of H2to CO
2.2.4 空速的影响
不同空速下催化剂性能随时间的变化结果如图12所示。由图12可知,随着空速增大催化剂稳定性降低,当空速从4 800 h-1增加至24 000 h-1,催化剂保持CO转化率100%的时间从120 h降低至36 h,也表明空速为4 800 h-1时催化剂性能稳定性较好。由前面分析可知,当空速增大时,催化剂表面Cα来不及向体相扩散,故在Ni表面沉积生成Cβ速率增大,故催化剂稳定性降低。

图12 空速对Ni/Al2O3催化剂性能影响Fig.12 The performance of Ni/Al2O3catalysts under differentGHSV
3 结 论
采用固相混合法制备催化剂方法简单,催化剂具有较好的还原性能和分散性。在甲烷化反应体系中随着压力和进料比(H2与CO体积比)增大,催化剂稳定性增强;随着空速增大,催化剂稳定性降低;升高温度会使催化剂表面积炭向更稳定形式沉积,从而催化剂稳定性增强。催化剂表面积炭的类型对催化剂活性和稳定性影响更大,当表面积炭的活性较高(Cα)或具有蠕虫状(Cv)形貌时,对催化剂活性影响不大,而无定形的胶质碳(Cβ)会使催化剂快速失活。
[1] Kopyscinski J, Schildhauer T J, Biollaz S M A. Production of synthetic natural gas (SNG) from coal and dry biomass-a technology review from 1950 to 2009 [J]. Fuel, 2010, 89(8):1763-1783.
[2] Rostrup-Nielsen J R, Pedersen K, Sehested J. High temperature methanation: Sintering and structure sensitivity [J]. Applied Catalysis A: General, 2007, 330:134-138.
[3] Nguyen T T M, Wissing L, Skjøth-Rasmussen M S. High temperature methanation: catalyst considerations [J]. Catalysis Today, 2013, 215:233-238.
[4] 张加赢, 辛 忠, 孟 鑫, 等. 基于MCM-41的镍基甲烷化催化剂活性与稳定性 [J]. 化工学报, 2014, 65(1):160-168. Zhang Jiaying, Xin Zhong, Meng Xin, et al. Activity and stability of nickel based MCM-41 methanation catalysts for production of synthetic natural gas [J]. Journal of Chemical Industry and Engineering, 2014, 65(1):160-168.
[5] Rostrup-Nielsen J R. Industrial relevance of coking [J]. Catalysis Today, 1997, 37(3):225-232.
[6] 刘 偲, 高明月, 李晨佳, 等. 工艺条件对镍基甲烷化催化剂积炭的影响 [J]. 精细石油化工, 2014, 31(2):1-6. Liu Cai, Gao Mingyue, Li Chenjia, et al. Effects of process conditions on the carbon deposition of the nickel-based methanation catalyst[J]. Speciality petrochemicals, 2014, 31(2):1-6.
[7] 刘化章, 董 昭, 陈珍珍, 等. 水蒸气浓度对合成气制甲烷Ni/γ-Al2O3催化剂稳定性影响 [J]. 天然气化工: C1化学与化工, 2013, 38(5):55-58. Liu Huazhang, Dong Zhao, Chen Zhenzhen, et al. Effect of water vapor concentration on stability of Ni/γ-Al2O3catalyst in syngas methanation [J]. Natural Gas Chemical Industry, 2013, 38(5):55-58.
[8] Pedersen K, Skov A, Rostrup-Nielsen J R. Catalytic aspects of high temperature methanation [J]. ACS Fuel Chem. Div Preprints, 1980, 25(2):89-100.
[9] Mirodatos C, Praliaud H, Primet M. Deactivation of nickel-based catalysts during CO methanation and disproportionation [J]. Journal of Catalysis, 1987, 107(2):275-287.
[10] Barrientos J, Lualdi M, Boutonnet M, et al. Deactivation of supported nickel catalysts during CO methanation [J]. Applied Catalysis A: General, 2014, 486:143-149.
[11] Bartholomew C H. Mechanisms of catalyst deactivation [J]. Applied Catalysis A: General, 2001, 212(1):17-60.
[12] Bartholomew C H. Carbon deposition in steam reforming and methanation [J]. Catalysis Reviews Science and Engineering, 1982, 24(1):67-112.
[13] Lu B, Kawamoto K. Preparation of the highly loaded and well-dispersed NiO/SBA-15 for methanation of producer gas [J]. Fuel, 2013, 103:699-704.
[14] Sun J F, Ge C Y, Yao X J, et al. Preparation of NiO/CeO2catalysts by solid state impregnation and their application in CO oxidation [J]. Acta Physico-Chimica Sinica, 2013, 29(11):2451-2458.
[15] Gao J, Jia C, Li J, et al. Ni/Al2O3catalysts for CO methanation: effect of Al2O3supports calcined at different temperatures [J]. Journal of Energy Chemistry, 2013, 22(6):919-927.
[16] Yang X, Gao G, Shi Q, et al. Impact of mesoporous structure of acid-treated clay on nickel dispersion and carbon deposition for CO methanation [J]. International Journal of Hydrogen Energy, 2014, 39(7):3231-3242.
[17] 陈珍珍, 董 昭, 王其刚, 等. 焙烧温度对Al2O3载体的性质及其负载的煤制合成气制代用天然气镍基催化剂性能的影响 [J]. 工业催化, 2013, 21(9):46-53. Chen Zhenzhen, Dong Zhao, Wang Qigang, et al. Efect of cal ination temperature on the properties of Al2O3support and catalytic performance of nickel-based catalyst for SNG from coal [J]. Industrial Catalysis, 2013, 21(9):46-53.
[18] Gao J, Wang Y, Ping Y, et al. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas [J]. RSC Advances, 2012, 2(6):2358-2368.
[19] 崔晓曦, 曹会博, 孟凡会, 等. 合成气甲烷化热力学计算分析 [J]. 天然气化工: C1化学与化工, 2012, 37(5):15-19. Cui Xiaoxi, Cao Huibo, Meng Fanhui, et al. Theromodynamic analysis for methanation of syngas [J]. Natural Gas Chemical Industry, 2012, 37(5):15-19.
[20] Bartholomew C H, Strasburg M V, Hsieh H Y. Effects of support on carbon formation and gasification on nickel during carbon monoxide hydrogenation [J]. Applied catalysis, 1988, 36:147-162.
[21] 李 霞, 杨霞珍, 唐浩东, 等. 载体对合成气制甲烷镍基催化剂性能的影响 [J]. 催化学报, 2011, 32(8):1400-1404. Li Xia, Yang Xiazhen, Tang Haodong, et al. Effects of supports on catalytic performance of nickel-based catalyst for methanation[J].Chinese journal of catalysis, 2011, 32(8):1400-1404.
[22] Ponec V. Some aspects of the mechanism of methanation and Fischer-Tropsch synthesis [J]. Catalysis Reviews Science and Engineering, 1978, 18(1):151-171.
[23] Forzatti P, Lietti L. Catalyst deactivation [J]. Catalysis today, 1999, 52(2):165-181.
[24] Baker R T K. Catalytic growth of carbon filaments [J]. Carbon, 1989, 27(3):315-323.
Stability for Performance of Nickel-Based Catalyst for Methanation under Different Reaction Conditions
Wei Xuemei, Tang Haodong, Li Xin, Liu Huazhang
Industry Catalysis Insitition of Zhejiang University of Technology, Hangzhou 310014, China
The methanation catalyst 20%Ni/Al2O3was prepared by solid state gridding method, and the effects of pressure, feeding ratio (volume ratio of H2to CO), space velocity, and temperature on the stability of the catalyst were studied. It was found that the catalyst prepared by the simple solid state gridding method had good dispersion and reduction performance. In the CO methanation reaction system, the stability of the catalyst was enhanced with the increase of temperature, pressure, and feeding ratio; the stability of the catalyst decreased with the increase of space velocity. The reasons that the catalyst deactivated were also discussed by the method of X-ray diffraction (XRD) and H2-temperature-programmed surface reaction (H2-TPSR). It was found that the catalyst was deactivated by Cβdeposited on the surface of the catalyst. Elevation of the temperature and pressure may help to shift the activated carbon species (Cα) on the surface of the catalyst to a more stable form, such as Ccor Cv, thereby enhanced the stability of the catalyst.
carbon monoxide; methanation catalyst; catalytic stability
TQ138.1+3
A
1001—7631 ( 2015 ) 05—0459—10
2015-03-23;
: 2015-06-23。
魏雪梅(1991—),女,硕士研究生;唐浩东(1975—),男,副教授,通讯联系人。E-mail:tanghd@zjut.edu.cn。
猜你喜欢
化工进展(2023年1期)2023-03-01
昆钢科技(2022年4期)2022-12-30
科学导报(2022年28期)2022-05-24
科学家(2022年3期)2022-04-11
发明与创新·大科技(2019年6期)2019-09-06
汽车维护与修理(2018年7期)2018-10-13
森林工程(2018年3期)2018-06-26
橡塑技术与装备(2016年14期)2016-02-24
化学反应工程与工艺(2015年1期)2015-04-16
燃气轮机技术(2014年4期)2014-04-16
