
Co-Mo/γ-Al2O3催化剂上噻吩与苯并噻吩加氢脱硫动力学
2015-01-07 03:37王雪杭段学志方向晨周兴贵
化学反应工程与工艺 2015年5期
杨 慧,王雪杭,段学志,方向晨,周兴贵
1.华东理工大学化学工程联合国家重点实验室,上海 200237;
2.抚顺石油化工研究院,抚顺 辽宁 113001
Co-Mo/γ-Al2O3催化剂上噻吩与苯并噻吩加氢脱硫动力学
杨 慧1,王雪杭1,段学志1,方向晨2,周兴贵1
1.华东理工大学化学工程联合国家重点实验室,上海 200237;
2.抚顺石油化工研究院,抚顺 辽宁 113001
采用固定床等温积分反应器,以不同组成的含硫化合物作为模型化合物,在消除催化剂内外扩散影响的基础上,考察了反应温度、苯并噻吩浓度、噻吩浓度以及硫化氢分压对加氢脱硫反应速率的影响,建立了苯并噻吩和噻吩单独加氢和共加氢的LHHW型反应动力学方程,并表明以真实的原料组成和在实际操作条件下进行动力学实验的重要性。
噻吩 苯并噻吩 加氢脱硫 动力学
成品油中的硫主要来自于催化裂化(FCC)汽油组分,因此,FCC汽油中硫的脱除是生产清洁汽油的关键。FCC汽油中的含硫化合物主要是噻吩(TF)、苯并噻吩(BT)及其衍生物[1-3]。目前,脱除这些化合物最有效的方法是加氢脱硫(HDS),最常用的催化剂是Co-Mo/Al2O3。
在对苯并噻吩和噻吩单独加氢脱硫的研究中,很多研究者认为含硫化合物与硫化氢吸附在同一活性位上,且这一活性位不同于氢气吸附的活性位,基于这一假设,研究者得到了L-H形式的动力学方程来表示这两种含硫化合物的反应速率[4-6]。然而,目前的研究大多关注的是含硫化合物单独加氢脱硫过程的动力学,对于两种含硫组分同时加氢脱硫过程的动力学目前尚未有研究,而这一研究对于理解FCC汽油的实际脱硫过程来说具有十分重要的意义,因为FCC汽油往往同时含有噻吩和苯并噻吩以及其衍生物等多种含硫化合物。
在研究汽油加氢脱硫反应的本征动力学时,需要考虑以下两个重要问题。第一,一种含硫化合物的加氢动力学是否具有代表性?在不同催化剂上,相同的含硫化合物会具有不同的加氢反应速率;不同的含硫化合物在同种催化剂上也会有不同的加氢反应速率。但在相同的催化剂上,不同的含硫化合物是否满足相同的动力学规律,即加氢速率与温度和浓度的数学关系是否不变?第二,在同一催化剂上,一种含硫化合物的加氢动力学规律是否会因为其他含硫化合物同时加氢过程而发生变化?对这些问题的回答能指导动力学实验,也能对不同含硫化合物的加氢机理以及这些过程的相互影响提供重要认识。
本工作以两种含硫化合物为研究对象,分别研究一种含硫化合物加氢脱硫和两种含硫化合物同时加氢脱硫时的速率与温度和浓度关系,讨论不同含硫化合物加氢脱硫动力学规律的差异,以及同时加氢脱硫时动力学规律的变化。
1 实验部分
1.1 实验装置及方法
由于汽油组分复杂,直接利用汽油进行实验将不利于产物的分析,因此实验中常利用模拟汽油替代实际汽油。具有代表性的模拟汽油应包含烷烃、芳香烃、烯烃和噻吩类含硫化合物。模拟油各组分及含量见表1。加氢脱硫动力学实验流程如图1所示。模拟汽油经高压恒流泵连续进入反应管,与氢气以及硫化氢混合后,在催化剂作用下发生加氢脱硫反应。反应产物进入气液分离器。未反应的氢气以及硫化氢经背压阀排出。反应液相产物采用气相色谱分析,色谱柱为OV-101石英毛细管柱,氢火焰(FID)检测器。
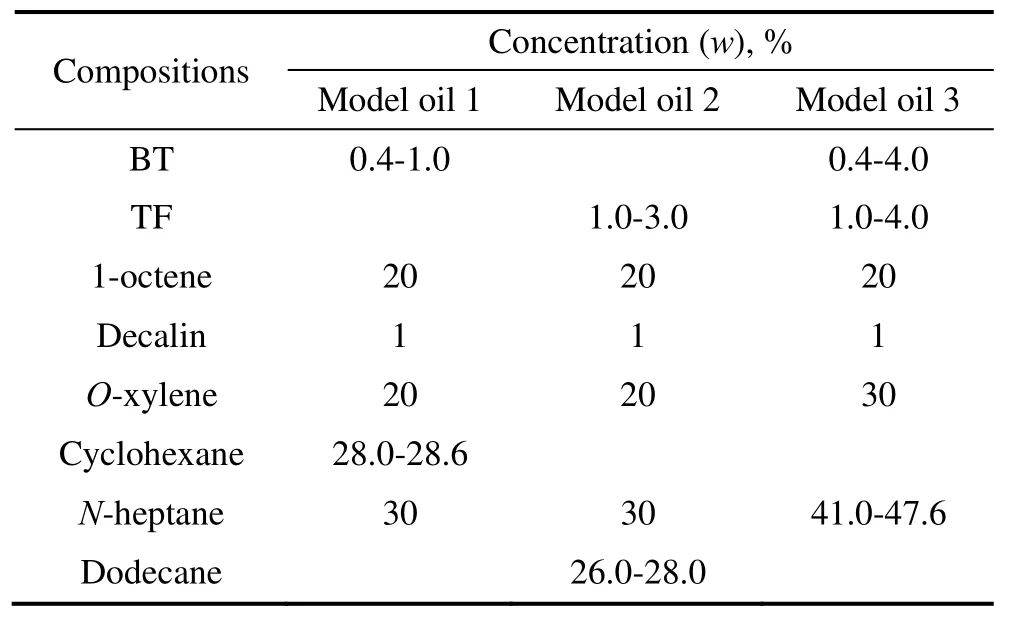
表1 模拟油各组分及含量Table 1 Composition and concentration of the model gasoline

图1 加氢脱硫实验装置Fig.1 Schematic diagram of experimental apparatus for the hydrodesulfurization
1.2 催化剂的预硫化与老化
在加氢脱硫之前,首先对催化剂进行预硫化处理,即在常压氮气保护下先升温至400 ℃,用10% H2S-H2混合气在50 mL/min流率下预硫化4 h。预硫化完成后,开启氮气吹扫整个管路,并降温至所需反应温度。继而在2 MPa和液体体积空速100 mL/(h·g)的条件下对催化剂进行老化处理。
1.3 内外扩散的排除实验
在2 MPa、340 ℃下,采用了三种不同颗粒粒径的催化剂进行内扩散排除实验,在不同的空速下进行外扩散的研究。研究表明当催化剂颗粒粒径小于40~60目,空速大于100 mL/(h·g)时,内外扩散的影响可以忽略。本研究的动力学实验均在排除了内外扩散的条件下进行。
1.4 动力学模型
苯并噻吩和噻吩加氢脱硫主要有两种可能反应路径,见图2和图3[7,8]。
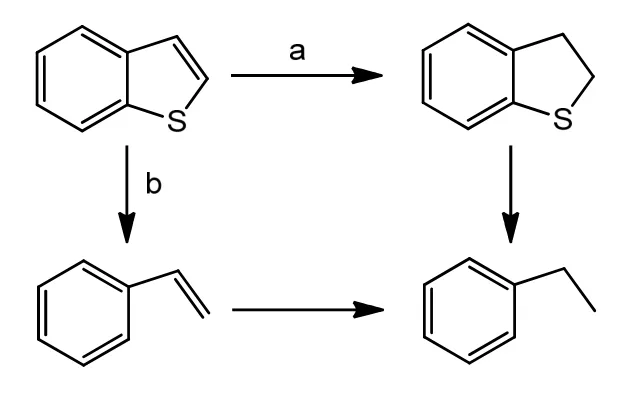
图2 BT加氢脱硫反应路径Fig.2 Reaction pathway of benzothiophene hydrodesulfurization
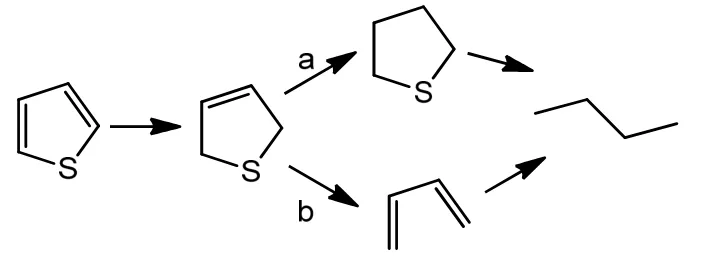
图3 TF加氢脱硫路径Fig.3 Reaction pathway of thiophene hydrodesulfurization
对苯并噻吩加氢脱硫时,路径a为主要反应路径;对噻吩加氢时,路径b是主要反应路径。他们的共同特点是先加氢、后脱硫,因此可用同一机理来分析。计A为噻吩或苯并噻吩,B为加氢的中间产物,即1,3-丁二烯或苯乙烯,C为加氢脱硫后的产物,即丁烷或乙苯。
单活性位机理[10]:A与H2S吸附在同一种活性位上,而H2吸附在另一种活性位上,A加氢为速率控制步骤,加氢产物在催化剂上的吸附为弱吸附。反应路径如下所示:

双活性位机理[11]:A加氢需要两个活性位,其余与单活性位机理相同。反应路径如下所示:
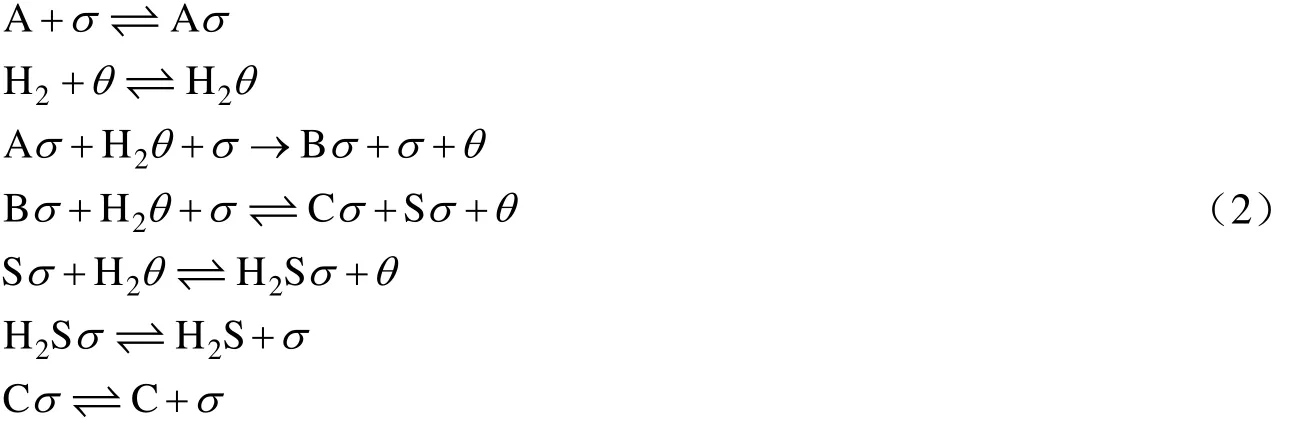
如果反应过程中氢分压保持不变(可通过使用大大过量的氢气实现),苯并噻吩和噻吩加氢分别有两种不同的反应动力学表达式,如表2所示。
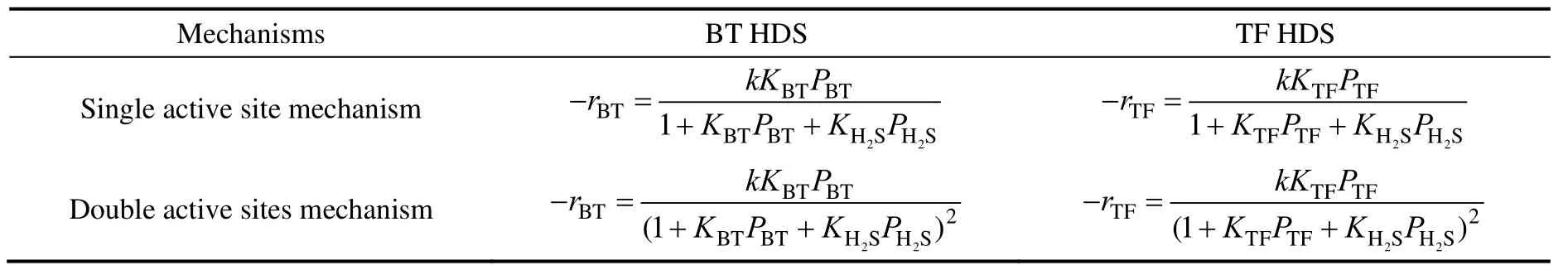
表2 苯并噻吩和噻吩单独加氢时的动力学方程Table 2 Kinetic equations of BT and TF hydrogenation separately
当模拟油中同时含有苯并噻吩和噻吩时,反应动力学方程主要有两种。由于苯并噻吩和噻吩分别有两种可能的机理,因此反应动力学方程有四种形式(见表3),即单活性位和双活性位的不同组合。

表3 苯并噻吩和噻吩共加氢时的动力学方程Table 3 Kinetic equations of BT hydrodesulfurization together with TF
为了验证上述机理,在四个温度水平下进行动力学实验,并在每个温度水平上改变进料浓度和空速。在反应条件下,模拟油充分汽化,反应器内发生气固催化反应。由于反应器直径小,气速大,可用平推流反应器模型进行数据处理。所有实验中的氢气大大过量。
2 结果与讨论
在排除了内外扩散的条件下,分别考察三种模拟油中苯并噻吩和噻吩的转化率随着反应温度、进料流率和进料浓度的变化情况。然后在平推流反应器模型基础上,通过非线性最小二乘法的lsqnonlin函数拟合实验数据从而求得模型参数[12]。
对于苯并噻吩加氢,采用双活性位动力学模型拟合出的H2S吸附平衡常数为零,这与H2S与苯并噻吩间存在竞争吸附作用的实验事实不符。与双活性位动力学模型相比,单活性位动力学模型在参数的物理意义上更合理,其动力学实验数据的拟合结果见图4。不同温度下的反应速率常数和吸附平衡常数列于表4。由此通过Arrhenius方程拟合得到的活化能为68.9 kJ/mol,通过Van't Hoff方程拟合得到的苯并噻吩与H2S的吸附热为分别为-74.4 kJ/mol与-57.9 kJ/mol。
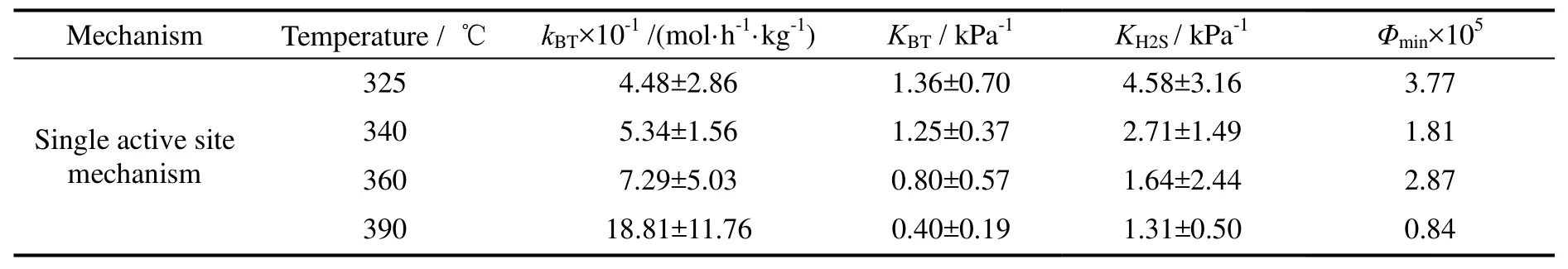
表4 BT加氢脱硫反应动力学模型参数Table 4 Kinetic model parameters of BT HDS separately
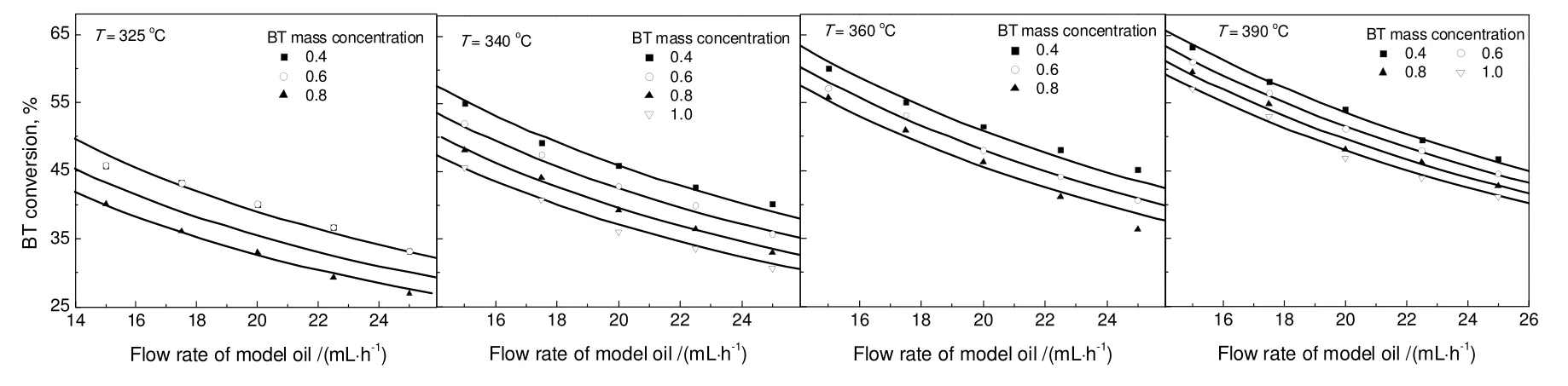
图4 不同温度下模拟油1中含硫化合物转化率实验数据与拟合结果的比较Fig.4 Fitting result of sulfur compounds conversion and the experimental data at different temperatures
对于噻吩加氢,通过单活性位模型拟合出来的KH2S等于0,这也与H2S与噻吩间存在竞争吸附作用的实验事实不符,双活性位模型在参数的物理意义上更具合理性,其动力学实验数据的拟合结果见图5。不同温度下的反应速率常数和吸附平衡常数列于表5中。由此通过Arrhenius方程拟合得到的活化能为29.7 kJ/mol,通过Van't Hoff方程拟合得到的噻吩与H2S的吸附热为分别为-11.1 kJ/mol与-18.7 kJ/mol。

表5 TF加氢脱硫反应动力学模型参数Table 5 Kinetic model parameters of BT HDS separately
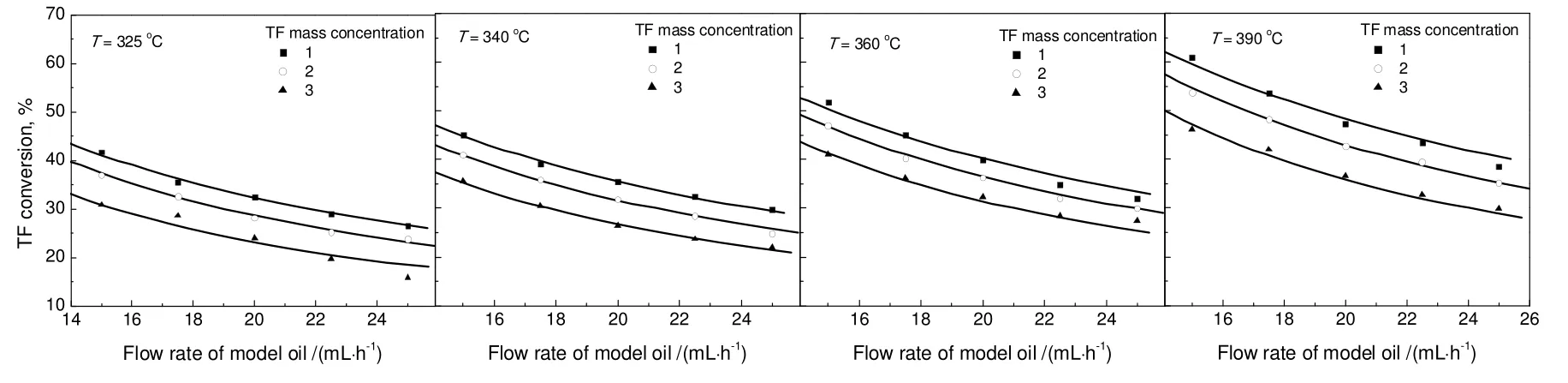
图5 不同温度下模拟油2中含硫化合物转化率实验数据的拟合结果Fig.5 Fitting result of sulfur compounds conversion and the experimental data at different temperatures
对苯并噻吩和噻吩同时加氢来说,反应机理1(苯并噻吩和噻吩加氢都只涉及一个活性位)与其他三种机理相比,无论从参数的物理意义还是参数的拟合效果上都更具合理性,其动力学实验数据的拟合结果见图6。不同温度下的反应速率常数和吸附速率常数见表6。由此得到的苯并噻吩和噻吩加氢活化能分别是83.2 kJ/mol和49.7 kJ/mol,苯并噻吩,噻吩以及H2S的吸附热分别是-65.6,-31.9,-40.8 kJ/mol。
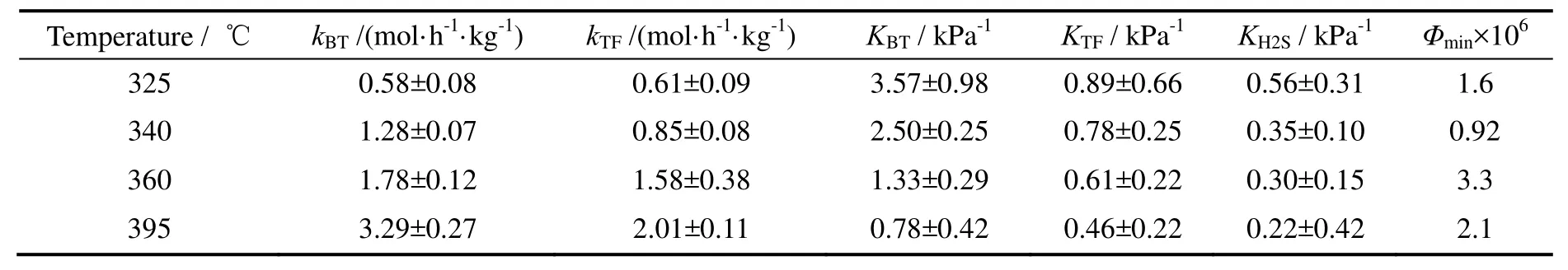
表6 BT-TF加氢脱硫反应动力学模型参数Table 6 Kinetic model parameters of BT hydrodesulfurization together with TF
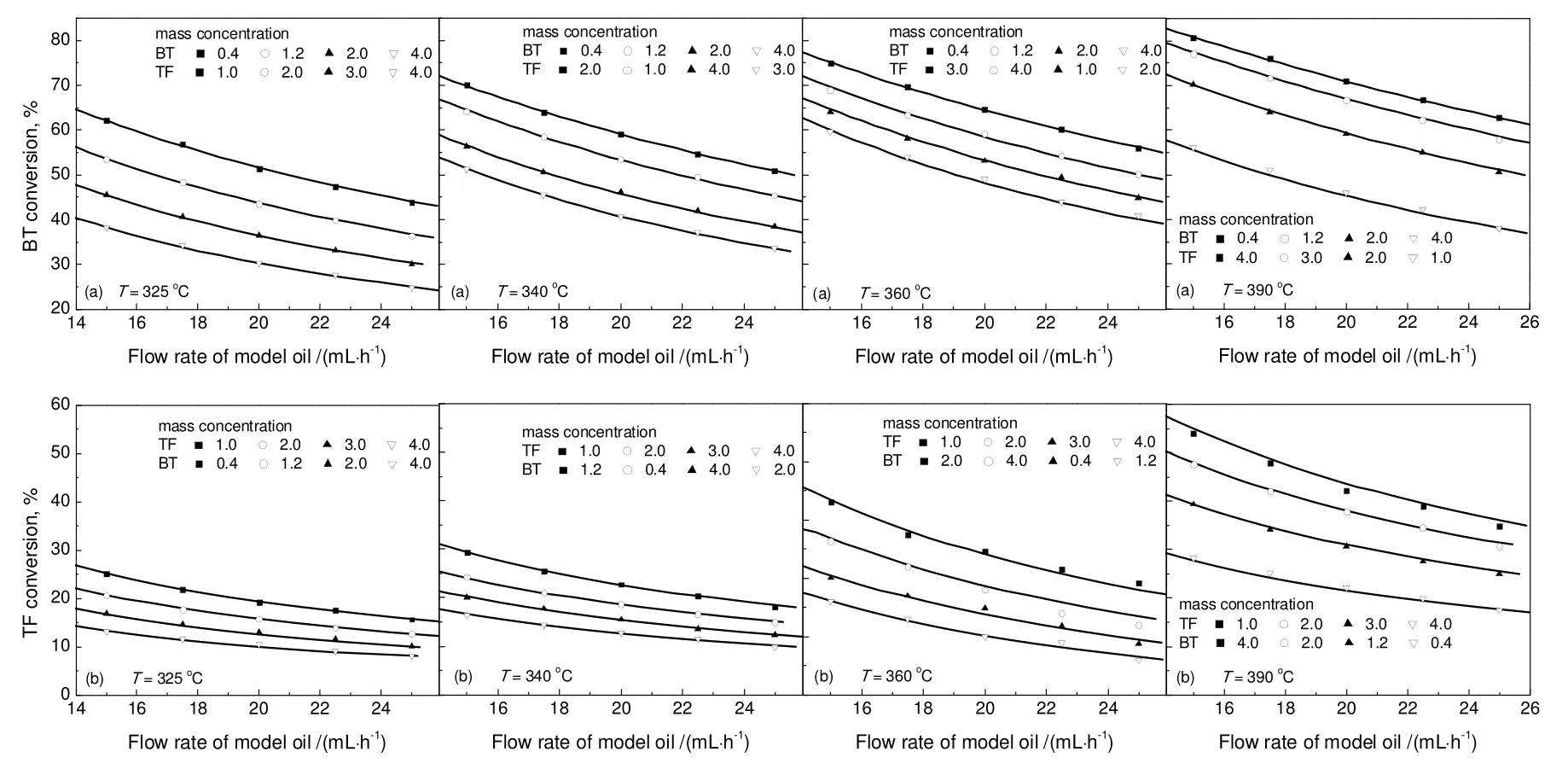
图6 不同温度下模拟油3中含硫化合物转化率实验数据与拟合结果的比较Fig.6 Fitting result of sulfur compounds conversion and the experimental data at different temperatures
以上结果表明,无论是独立加氢还是共加氢,苯并噻吩的活化能都大于噻吩的活化能,而且共加氢的活化能高于独立加氢。动力学计算表明,在相同的温度和组分(苯并噻吩或噻吩)浓度下,单独加氢时苯并噻吩或噻吩的反应速率要大于共加氢时苯并噻吩或噻吩的反应速率。单独加氢时反应发生在最有利于其转化的活性位和路径上(自由能下降速率最大),而共加氢时发生在同时有利于两种物质转化的活性位和反应路径上;苯并噻吩与噻吩加氢的活化能有所提高就是兼顾苯并噻吩与噻吩共加氢的结果。
比较苯并噻吩与噻吩单独加氢和共加氢的吸附热,可看出在单独加氢变为共加氢时,强吸附变弱而弱吸附变强。由于吸附质之间的相互作用,通常一个物质的吸附会削弱另一个物质的吸附,但前提是这两种物质在反应中吸附在相同的活性位上[12]。而独立加氢和共加氢,与反应动力学相关的吸附不一定发生在相同的活性位上。这也可以从噻吩在单独加氢和共加氢时反应机理上的变化(从双活性位机理变为单活性位机理)上看出。硫化氢与噻吩和苯并噻吩之间存在竞争吸附,因此噻吩和苯并噻吩吸附位的变化也将影响硫化氢的吸附(包括吸附位、吸附构型和吸附热)。由此可以看出不同的含硫化合物在相同的催化剂上有不同的动力学规律。此外,加氢时一个含硫化合物的加氢动力学规律会受到其他含硫化合物加氢反应的影响。
LHHW动力学方法建立在Langmuir吸附基础上,从原理上除要求吸附为单层吸附外,还要求同种吸附质在固体表面所有吸附位的吸附能力相同,且不受该物质吸附覆盖度以及其他物质共吸附过程的影响。实际催化剂表面存在很大程度的不均匀性,如在Co-Mo/γ-Al2O3催化剂上,含硫化合物存在不同的吸附方式,即通过S原子垂直吸附以及芳香环的平行吸附[13]。对应这两种吸附方式,含硫化合物有直接脱硫和加氢脱硫两种反应机理。对某一含硫物种,两种反应机理的竞争性与该物种和其他物种的表面覆盖度有关。
研究表明,一个加氢脱硫反应的LHHW动力学无论从形式还是从参数上都会受其他反应和吸附的影响。上述分析表明,不能通过单个含硫化合物的加氢动力学构建多个含硫化合物共加氢的动力学。这里也可推断,在远离工业操作条件下建立的LHHW动力学也不具备工业应用价值。
3 结 论
本工作研究了苯并噻吩与噻吩单独加氢和共加氢的动力学。苯并噻吩单独加氢的决速步骤涉及一个苯并噻吩吸附位,噻吩单独加氢的决速步骤涉及两个噻吩吸附位;而在共加氢中,苯并噻吩和噻吩加氢的决速步骤都只涉及一个苯并噻吩或噻吩活性位。由于苯并噻吩、噻吩和H2S之间存在竞争吸附,且不同吸附质之间的吸附存在相互影响,采用LHHW方法建立动力学必须以真实的原料组成和实际操作条件为基础。
符号说明
ki—— 含化合物i的加氢脱硫表观反应速率常数,mol/(h·kg)Ki——化合物i的吸附平衡常数,kPa-1
-ri—— 含化合物i的加氢脱硫反应速率,mol/(h·kg)Φmin——最小残差平方和,[mol/(h·kg)]2
[1] Kaufmann T G, Kaldor A, Stuntz G F, et al. Catalysis science and technology for cleaner transportation fuels [J]. Catalysis Today, 2000, 62(1):77-90.
[2] Brunet S, Mey D, Perot G, et al. On the hydrodesulfurization of FCC gasoline: a review [J]. Applied Catalysis A: General, 2005, 278(2):143-172.
[3] Babich I V, Moulijn J A. Science and technology of novel processes for deep desulfurization of oil refinery streams: a review [J]. Fuel, 2003, 82(6):607-631.
[4] Kilanowski D R, Gates B C. Kinetics of hydrodesulfurization of benzothiophene catalyzed by sulfided Co-Mo/Al2O3[J]. Journal of Catalysis, 1980, 62(1):70-78.
[5] Satterfield C N, Roberts G W. Kinetics of thiophene hydrogenolysis on a cobalt molybdate catalyst [J]. AIChE Journal, 1968, 14(1):159-164.
[6] Lee H C, Butt J B. Kinetics of the desulfurization of thiophene: reactions of thiophene and butane [J]. Journal of Catalysis, 1997, 49(3):320-331.
[7] Yao X Q, Li Y W, Jiao H J. Mechanistic aspects of catalyzed benzothiophene hydrodesulfurization. a density functional theory study [J]. Journal of Molecular Structure: THEOCHEM, 2005, 726(1-3):67-80.
[8] Tanaka K I, Okuhara T. Regulation of intermediates on sulfide nickel and MoS2catalysts [J]. Catalysis Reviews-Science and Engineering, 1997, 15(2):249-292.
[9] Singhal G H, Espino R L, Sobel J E, et al. Hydrodesulfurization of sulfur heterocyclic compounds: kinetics of dibenzothiophene [J]. Journal of Catalysis, 1981, 67(2):457-468.
[10] Broderick D H, Gates B C. Hydrogenolysis and hydrogenation of dibenzothiophene catalyzed by sulfide CoO-MoO3/Al2O3: the reaction kinetics [J]. AIChE Journal, 1981, 27(4):663-673.
[11] 黄华江. 使用化工计算机模拟 [M]. 北京: 化学工业出版社, 2012:202-203.
[12] Yang M L, Zhu J, Zhu Y, et al. Tuning selectivity and stability in propane dehydrogenation by shaping Pt particles: a combined experimental and DFT study [J]. Journal of Molecular Catalysis A: Chemical, 2014, 395(?):329-336.
[13] Daage M, Chianelli R R. Structure-function relations in molybdenum sulfide catalysts: the rim-edge model [J]. Journal of Catalysis, 1994, 194(2):414-427.
Kinetics of Thiophene and Benzothiophene Hydrodesulfurization over Co-Mo/γ-Al2O3Catalyst
Yang Hui1, Wang Xuehang1, Duan Xuezhi1, Fang Xiangchen2, Zhou Xinggui1
1. State Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China; 2. Fushun Research Institute of Petroleum and Petrochemicals, SINOPEC, Fushun 113001, China
Hydrodesulfurization of thiophene and/or benzothiophene was carried out on Co-Mo/γ-Al2O3catalyst in an isothermal integral reactor under different reaction temperatures, sulfur-containing compound concentrations and hydrogen sulfide partial pressures. LHHW reaction kinetic equation for hydrodesulfurization of thiophene alone or together with benzothiophene was established. The importance of kinetic study using real composition of raw materials under actual industrial operating conditions has been demonstrated.
thiophene; benzothiophene; hydrodesulfurization; kinetics
TE624.9+3; TQ032.4l
A
1001—7631 ( 2015 ) 05—0400—07
2015-03-31;
: 2015-05-12。
杨 慧(1990—),女,硕士研究生;周兴贵(1966—),男,教授,通讯联系人。E-mail:xgzhou@ecust.edu.cn。
国家自然科学基金(91434117)。
猜你喜欢
建材发展导向(2021年14期)2021-08-23
西南石油大学学报(自然科学版)(2021年3期)2021-07-16
环境保护与循环经济(2021年12期)2021-03-16
中国煤层气(2019年2期)2019-08-27
环境与可持续发展(2017年2期)2017-04-06
当代化工研究(2016年1期)2016-03-16
中国资源综合利用(2016年7期)2016-02-03
合成化学(2015年10期)2016-01-17
应用化工(2014年9期)2014-08-10
火炸药学报(2014年1期)2014-03-20
