
基于分子印迹磁性复合材料的基质分散-固相萃取/液相色谱法测定海水中的氯酚类污染物
2015-01-01 02:34王新鑫谢晟瑜刘芳伶沈昊宇夏清华
分析测试学报 2015年11期
王新鑫,杨 军,谢晟瑜,陈 扬,刘芳伶,沈昊宇*,夏清华
(1.浙江大学 宁波理工学院,浙江 宁波 315100;2.湖北大学 有机化工新材料湖北省协同创新中心有机功能分子合成与应用教育部重点实验室,湖北 武汉 430062)
氯酚类化合物(CPs),如:氯酚(CP)、二氯苯酚(DCP)、三氯苯酚(TCP)、四氯酚(TeCP)和五氯酚(PCP)等具有较强的生物蓄积毒性和“三致”效应,被美国环境署(EPA)和中国国家环保总局列入优先控制污染物的黑名单[1-2]。随着人们海洋环境保护意识的加强,海洋环境中痕量酚类环境污染物的检测与防治已成为重点研究方向之一。目前,氯酚类化合物的检测方法主要有气相色谱法[3-4]、液相色谱法[5-6]和毛细管电泳法[7-8]等。然而,海水基体复杂,氯酚类化合物含量处于痕量水平[5],往往需富集净化后才能准确测定。现有的样品富集方法存在吸附容量小、富集能力差、不易实现固液分离等不足。磁性复合材料因具有良好的磁响应性,在外磁场作用下可以实现快速、有效的固液分离而成为国内外研究的重要方向之一[9]。已有研究以此为基础建立了基质分散-磁性固相萃取(dMSPE)技术对样品进行预处理[10]。dMSPE技术是以磁性或可磁化的材料作富集材料基质的一种固相萃取技术,在磁固相萃取过程中,磁性富集材料被添加到样品溶液或悬浮液中用以吸附目标分析物,并在外部磁场作用下分离;避免了普通固相萃取技术过柱操作繁琐、吸附柱易堵塞、重复性差等问题,具有良好的应用前景。
分子印迹聚合物(MIP)是一类新兴的富集材料[11-12],在复杂基质中目标物的富集与净化方面具有良好的应用前景。通常将模板分子(Template molecular)和功能单体(Functional monomer)先通过共价或非共价键作用,形成主客体配合物;然后通过聚合反应得到聚合物;最后用适当的溶剂将模板分子洗脱。所得的聚合物拥有特异性的结合位点,对模板分子的功能基团、分子尺寸、空间结构等具有记忆功能,可以根据预定的选择性和高识别性进行分子识别。然而正是由于其对底物的高选择性识别,导致结构与性质相近的同系物难以实现同时测定,因此模板分子的选择对于同系物的同时分析至关重要。本文以五氯酚为分子印迹模板,利用氢键使其与氨基结合,然后偶联到磁性纳米Fe3O4材料表面,洗脱五氯酚后得到分子印迹氨基功能化磁性复合材料(nFe3O4@MIPNH2-polymer)。在此基础上建立了一种基质分散-磁性固相萃取/液相色谱(dMSPE-HPLC)方法用以富集检测海水中痕量的氯酚,实现了氯酚类化合物的高选择性识别和富集,富集后的复合材料在外加磁场作用下可实现快速磁分离。该方法具有快速、高效、灵敏、环境友好等优点。
1 实验部分
1.1 试剂与仪器
4-氯酚(4-CP)、2,4-二氯酚(2,4-DCP)、2,4,6-三氯苯酚(2,4,6-TCP)、2,3,4,6-四氯苯酚(2,3,4,6-TeCP)、五氯苯酚(PCP)(分析纯,阿拉丁试剂有限公司);二乙烯苯(DVB)、甲基丙烯酸缩水甘油酯(GMA)(分析纯,Fluke试剂有限公司);三氯化铁(FeCl3·7H2O)、四乙烯五胺(TEPA)、乙二醇(EG)、聚乙二醇400(PEG400)、乙酸钠、乙酸铵、2,2’-偶氮双(异丁腈)(ABIN)(分析纯,国药集团化学试剂有限公司)。甲醇(色谱纯,Merck试剂有限公司),其余试剂均为分析纯。模拟海水样品按照文献配制[13],组成为:NaCl,24.53 g/L;MgCl2,5.20 g/L;Na2SO4,4.09 g/L;CaCl2,1.16 g/L;KCl,0.695 g/L;NaHCO3,0.201 g/L;KBr,0.101 g/L;H3BO3,0.027 g/L;SrCl2,0.025 g/L;NaF,0.039 g/L。
SM-6700F扫描电子显微镜(SEM);日立H-7650透射电子显微镜(TEM);Bruker D8 Advance XRD分析仪;Lake Shore 7410振动样品磁强计测量计;Flash-1112型有机元素分析仪(EA);傅立叶变换红外光谱仪(NEXUS-470);大连伊利特液相色谱仪;P230Ⅱ高压恒流泵。
1.2 溶液的制备
分别准确称取0.05 g各氯酚类化合物溶于100 mL甲醇中,配成500 mg/L的储备液;移取一定体积的储备液用模拟海水稀释定容成工作液,当天配制。实际海水样品采自宁波近海。
nFe3O4@MIPNH2-polymer中Fe的含量采用邻二氮菲比色法测定,Fe3O4含量根据式(1)计算得到:

式中CFe为根据比色法实测的消解定容后溶液中Fe的质量浓度(mg/L);V为消化后溶液的定容体积(mL);72.36%为Fe3O4中Fe的百分含量;m为nFe3O4@MIPNH2-polymer的质量(mg)。
1.3 基质分散-磁性固相萃取(dMSPE)处理方法
移取50 mL样品溶液于150 mL平底烧瓶中,加入20 mg nFe3O4@MIPNH2-polymer,308 K下恒温振荡10 min,磁分离,弃去上层溶液。然后加入1 mL 1%NaOH的甲醇溶液,振荡4 min后进行脱附,磁分离,吸取上层清液,氮吹至干,以200 μL 5%的HCl-甲醇溶液溶解,过0.2 μm滤膜,在液相色谱仪上进样分析。
1.4 液相色谱测定条件
采用伊利特 C8反相液相色谱柱(250 mm×4.6 mm×5.0 μm)分离,以体积比为70∶30的甲醇-5 mmol/L乙酸铵水溶液为流动相,流速1 mL/min,进样量20 μL,UV230Ⅱ紫外检测器(UVD)在230 nm处检测。
2 结果与讨论
2.1 nFe3O4@MIPNH2-polymer的制备
分子印迹氨基功能化磁性高分子材料(nFe3O4@MIPNH2-polymer)按改进方法合成[14],其制备过程如图1所示。先采用溶剂热法得到磁性纳米Fe3O4,进一步通过沉淀聚合法得到环氧基功能化的Fe3O4-高分子材料Fe3O4@P(GMA-co-DVB)(图1A);五氯酚(PCP)与四乙烯五胺(TEPA)通过氢键结合得到PCP-TEPA模板分子(图1B);进一步与Fe3O4@P(GMA-co-DVB)开环键合得到氨基功能化高分子材料(图1C);洗脱模板分子PCP后得到分子印迹氨基功能化磁性高分子材料nFe3O4MIP@NH2-polymer(图1D)。
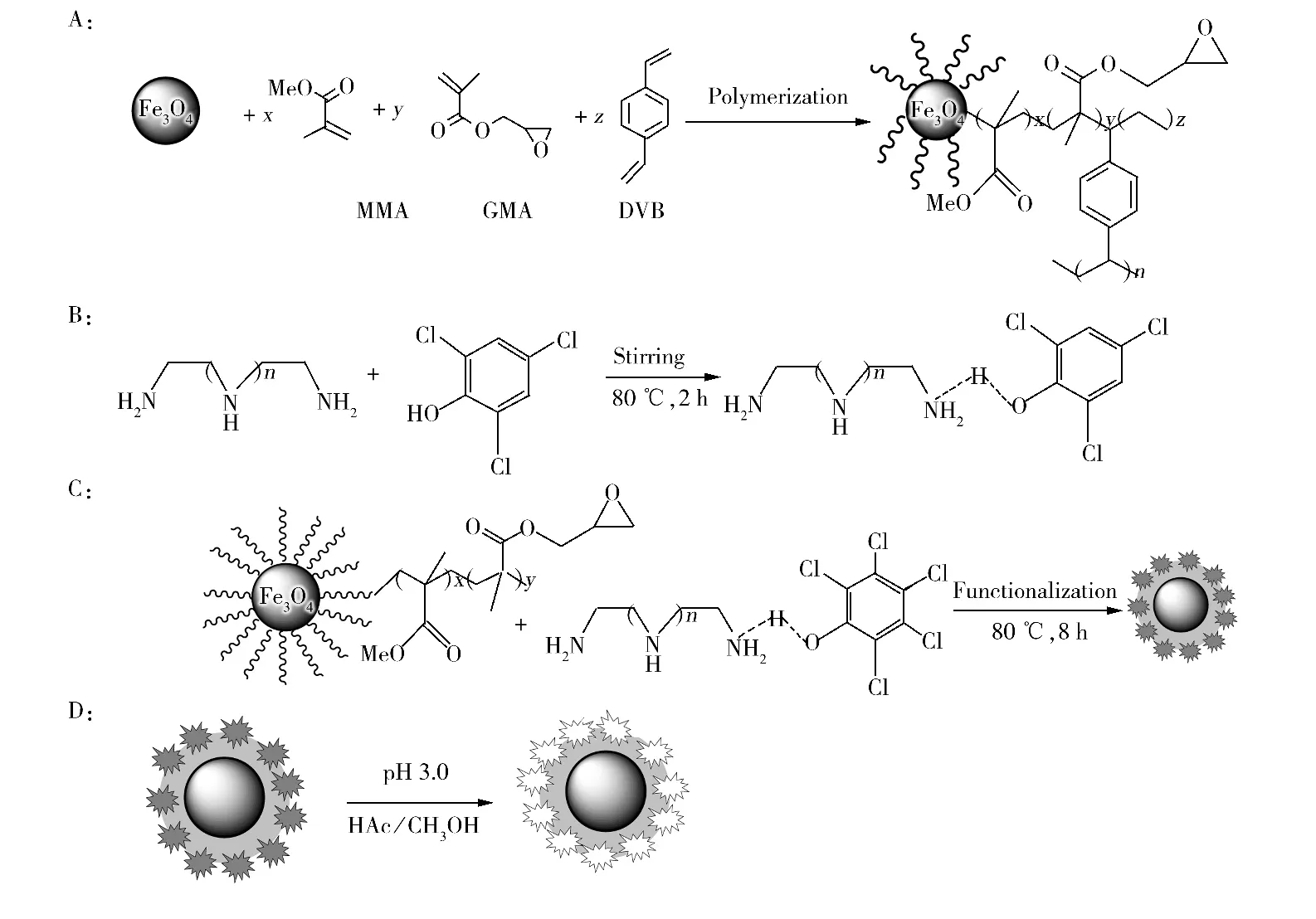
图1 纳米Fe3O4@分子印迹氨基功能化高分子磁性复合材料(nFe3O4@MIPNH2-polymer)的合成Fig.1 Synthesis of pentachlorophenol-imprinted amino-functionalized nano-Fe3O4-polymer magnetic composite material(nFe3O4@MIPNH2-polymer)
2.2 nFe3O4@MIPNH2-polymer的表征
图2A和B分别为nFe3O4@MIPNH2-polymer的SEM图和TEM图。由SEM图可知,nFe3O4@MIPNH2-polymer具有均匀的球形结构;由TEM图可知,nFe3O4@MIPNH2-polymer为典型的壳核结构,平均粒径约为800 nm。深色部分为Fe3O4磁核,平均粒径约为200 nm;浅灰色部分为高分子层,均匀分布在磁核外层,厚度约为300 nm。

图2 nFe3O4@MIPNH2-polymer的 SEM(A)和 TEM(B)图Fig.2 SEM(A)and TEM(B)images of nFe3O4@MIPNH2-polymer
图3为nFe3O4@MIPNH2-polymer的XRD图谱,与纯的nFe3O4相比,出现了Fe3O4的6个典型衍射峰,其2θ角分别位于30.1°(220),35.5°(311),43.1°(400),53.4°(422),57.0°(511)和62.6°(440)。可见 nFe3O4@MIPNH2-polymer的晶相未发生变化,保持了Fe3O4的尖晶石结构。图4为 nFe3O4@MIPNH2-polymer的磁滞回线(VSM);可知其磁化强度为32.6 emu/g,说明nFe3O4@MIPNH2-polymer的磁响应良好,能够在磁场下实现良好分离。nFe3O4@MIPNH2-polymer的元素分析实验值(%)为:H:1.95,C:48.24,N:10.57,O:24.22,经式(1)计算得到Fe3O4的实验值(%)为:15.04。材料的红外光谱主要特征吸收峰:3 400~3 500 cm-1和约1 575 cm-1,分别归属为 N—H的伸缩振动和变形振动;约1 750 cm-1处归属为羰基的伸缩振动,约580 cm-1处归属为Fe3O4的特征峰。表明氨基功能化高分子已成功修饰到Fe3O4表面。
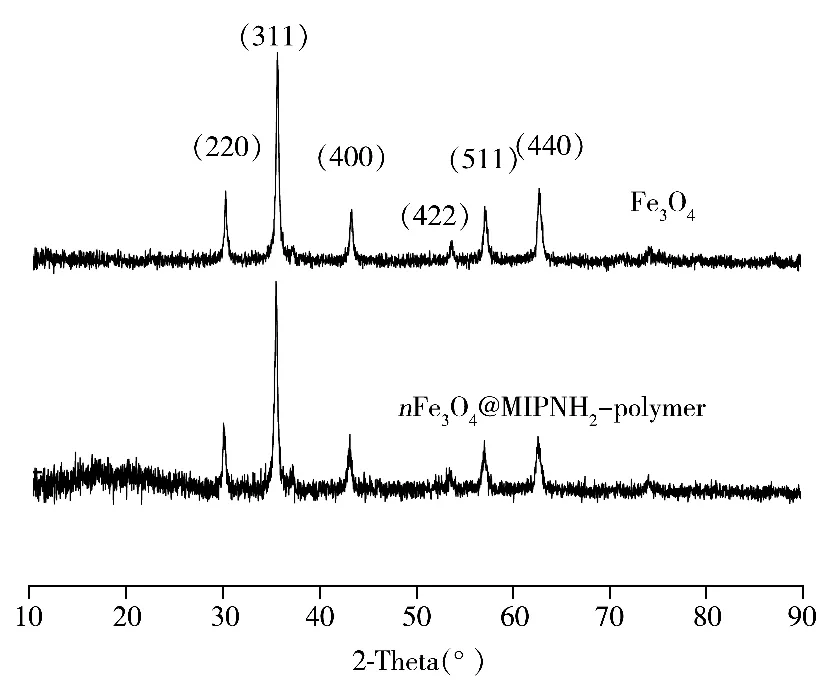
图3 Fe3O4和nFe3O4@MIPNH2-polymer的XRD图谱Fig.3 XRD spectra of nFe3O4and nFe3O4@MIPNH2-polymer
2.3 富集条件的优化
2.3.1 pH值的优化 溶液pH值显著影响富集材料对目标物的固相萃取性能。配制5种氯酚(CPs)混合溶液的浓度为2.0 μg/L,分别调节溶液pH值为2.0~10.0,加入20 mg富集材料nFe3O4@MIPNH2-polymer,于308 K温度下恒温振荡1 h,经磁分离后,取上层清液,采用HPLC检测5种CPs的浓度,结果如图5所示。由图可知,nFe3O4@MIPNH2-polymer对CPs的萃取效率受溶液pH值的影响较大,其萃取效率随着溶液pH值的增大而增大;当4.0≤pH≤8.5时,富集材料对CPs的萃取率达到最大,且出现平台;当pH>8.5时,继续增大溶液的pH值,萃取量反而减小。可能的原因是:当pH<4.0时,nFe3O4@MIPNH2-polymer表面的氨基主要以—NH+3的形式存在,与CPs分子之间难以形成氢键,主要以π-π相互作用,所以萃取率较低;pH 4.0~8.5时,富集材料表面氨基主要以—NH2存在,能与氯酚分子间形成氢键(—NH···O),nFe3O4@MIPNH2-polymer的高分子层中苯环与氯酚的苯环间存在π-π相互作用,所以萃取率较大;当pH>8.5时,CPs分子中羟基以—O-形式存在,不利于氢键的形成,所以萃取率逐渐降低。因此,为了获得高的提取效率,选择溶液的pH值为4.0~8.5。因实际海水样品的pH值为7.6~8.2,后续实验无需调节样品pH值,可以直接对实际海水样品进行测定。
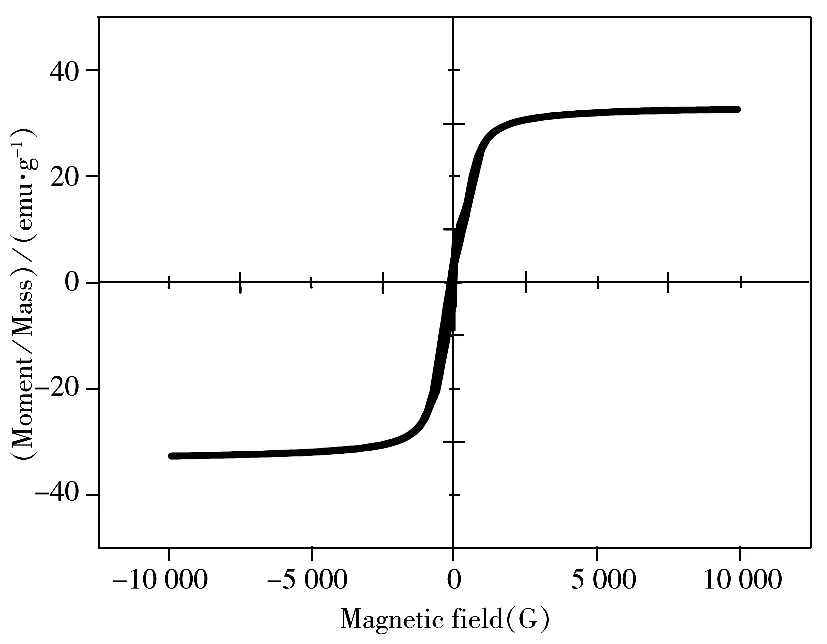
图4 nFe3O4@MIPNH2-polymer的VSM曲线Fig.4 VSM curve of nFe3O4@MIPNH2-polymer
2.3.2 萃取时间的优化 配制5种CPs混合溶液的浓度为2.0 μg/L,加入富集材料nFe3O4@MIPNH2-polymer 20 mg,于308 K温度下恒温振荡,分别控制萃取时间为1~20 min,以考察萃取时间对5种CPs固相萃取性能的影响。萃取结束后磁分离,对上清液中CPs的含量进行分析,结果显示,随着萃取时间的延长,5种CPs的萃取率逐渐增大,当萃取时间超过8 min后,萃取率达到最大。此后,进一步延长萃取时间,化合物的萃取率基本保持不变。为保证萃取完全,本实验选择最优萃取时间为10 min。
图5 溶液pH值对nFe3O4@MIPNH2-polymer固相萃取CPs的影响Fig.5 Effect of pH value on extraction efficiency of nFe3O4@MIPNH2-polymer to CPs
2.3.3 洗脱剂的选择 分别选择甲醇、2%氨水甲醇溶液、1%NaOH甲醇溶液作为5种CPs的洗脱剂,控制溶液体积为1 mL,恒温振荡20 min,测定结果如图6所示。由图6可知,当以甲醇作为洗脱剂时,2,4,6-TCP,2,3,4,6-TeCP和PCP的回收率可达80%以上,而另外2种CPs的回收率较低(<80%)。可能的原因是:2,4,6-TCP,2,3,4,6-TeCP和PCP苯环上的取代基团较多,酚羟基较难与nFe3O4@MIPNH2-polymer之间形成氢键作用力,而主要通过π-π相互作用力与高分子中的苯环相结合,甲醇能将其破坏,所以三者的脱附回收率较高;相反,4-CP和2,4-DCP与富集材料的氨基之间存在氢键作用力,甲醇难以将其破坏,所以回收率较低。当采用2%氨水甲醇作为洗脱剂时,2,4-DCP的回收率明显提高,可能是碱性条件不利于氢键的形成,但4-CP(73.2%)的回收率并未显著改善,可能是因为其沸点相对较低,在洗脱剂氮吹挥干过程中4-CP有损失。为此,实验采用1%NaOH甲醇溶液,将4-CP转化成钠盐,以减小挥干过程中的损失,提高脱附回收率。结果显示,当采用1%NaOH甲醇作为洗脱剂时,4-CP的脱附回收率较为理想(>95%),且其余4种氯酚的回收率也较高。因此,实验选择洗脱剂为1%NaOH甲醇。
2.3.4 洗脱剂体积与脱附时间的优化 分别以0.5,1,2,3 mL的1%NaOH甲醇为洗脱剂考察其体积对CPs回收率的影响。结果表明,当洗脱剂的体积≥1 mL时,5种CPs的回收率均高于90%,且随着洗脱剂体积的增加,回收率无明显增加;而增加洗脱剂的体积,不仅溶剂用量增加,且后续氮吹的时间也较长,因此,选择1 mL 1%NaOH甲醇为洗脱剂。
进一步考察了脱附时间的影响。结果显示,随着脱附时间的延长,5种CPs的脱附率逐渐增大,2,4,6-TCP,2,3,4,6-TeCP和PCP达最大脱附率所需的时间为2 min,4-CP和2,4-DCP达最大脱附率所需时间为4 min。综合考虑,选择4 min作为5种CPs的脱附时间。
2.3.5 海水中主要无机盐的影响 海水是一种非常复杂的多组分水溶液。海水中各种元素均以一定的物理化学形态存在,主要成分(大量、常量元素)为浓度大于1.0×103mg/kg的成分,属于此类的阳离子有Na+,K+,Ca2+,Mg2+和Sr2+,阴离子有Cl-,SO2-4,Br-,HCO-3(CO2-3),F-,以及以分子形式存在的H3BO3,其总和占海水盐分的99.9%[13]。本文通过添加不同浓度的NaCl,Na2SO4,KCl,NaF,KBr,CaCl2,NaHCO3,Na2CO3,MgCl2和SrCl2考察了基体中添加剂的影响。结果表明,上述物质的含量低于5%时对加标浓度为50 ng/L CPs的回收率基本无影响。
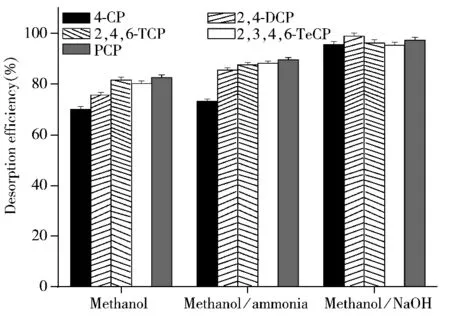
图6 洗脱剂种类对CPs脱附效率的影响Fig.6 Effect of desorption solvent on desorption efficiency to CPs
2.4 线性关系、检出限及定量下限
在优化条件下,考察5种CPs的线性范围,结果如表1所示。富集后5种CPs在1~5 000 ng/L浓度范围内呈良好的线性关系,相关系数(r2)均大于0.998 9,而富集前5种CPs的线性范围为0.4~1 000 μg/L。以富集前后标准曲线的斜率比计算富集倍数[15-16],发现本方法所得富集倍数可达222.2~245.7倍。以3倍信噪比(S/N=3)和10倍信噪比(S/N=10)分别计算检出限(LOD)和定量下限(LOQ),得5种CPs的LOD为0.18~1.20 ng/L,LOQ为0.6~4.0 ng/L。
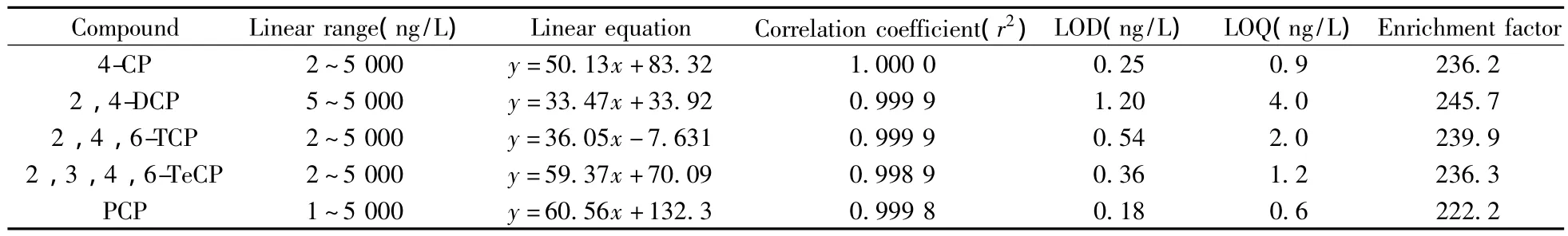
表1 5种CPs的线性关系、检出限(LODs)、定量下限(LOQs)与富集倍数Table 1 Linear relationships,LODs,LOQs and enrichment factors of 5 kinds of CPs
2.5 回收率与精密度
以不含待测物的模拟海水样品为试剂空白,分别加入5种CPs进行低、中、高(2.0,10.0,50.0 ng/L)3个浓度水平的加标回收实验(表2)。5种CPs在3种加标水平下的回收率分别为86.5%~97.5%,92.8%~98.2%和88.8%~98.8%,日内相对标准偏差(RSD)为0.8%~7.2%,日间(7 d)RSD为1.2%~8.6%。
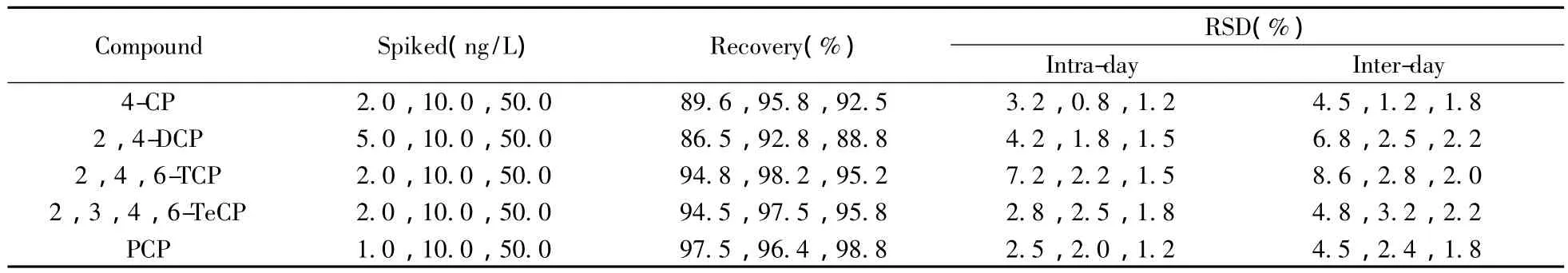
表2 5种CPs在模拟海水样品中的回收率与相对标准偏差(n=5)Table 2 Recoveries and RSDs of five CPs in synthesized sea water samples(n=5)
2.6 实际样品的检测
采用本方法对3种实际样品中的CPs进行检测,发现1种样品中含有2,4,6-TCP,浓度为8.6 ng/L。图7为空白模拟海水样品、空白模拟海水样品加标及实际样品的HPLC图谱,由图可见本文所建立的dMSPE方法可以有效去除海水中的基体干扰,达到富集CPs的目的。与已有检测CPs的方法相比(表3),本方法具有操作简单、检出限低等优点。
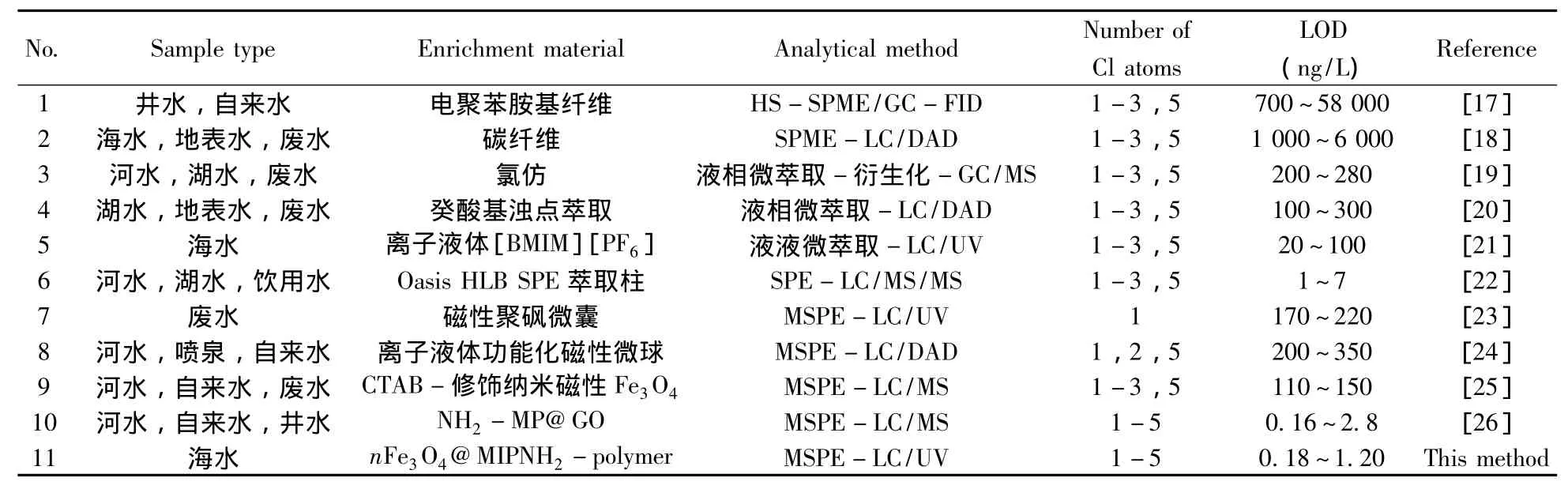
表3 水样中CPs的检测方法比较Table 3 Comparison of analytical methods of CPs in water samples
3 结论
本文建立了检测海水中5种氯酚类化合物(CPs)残留量的基质分散-磁性固相萃取/液相色谱分析方法(dMSPE-HPLC)。样品经纳米Fe3O4分子印迹氨基功能化高分子磁性复合材料(nFe3O4@MIPNH2-polymer)富集,基质分散-磁性固相萃取,通过磁分离后进行液相色谱法检测。5种CPs在1~5 000 ng/L浓度范围内具有良好的线性关系,相关系数(r2)大于0.998 9,检出限(LOD)为0.18~1.20 ng/L,定量下限(LOQ)为0.6~4.0 ng/L,平均回收率为 86.5%~98.8%,RSD为0.8%~8.6%。该方法前处理快速简便,无需特别装置,试剂用量少,对环境污染小,可以实现5种CPs化合物的同时测定。
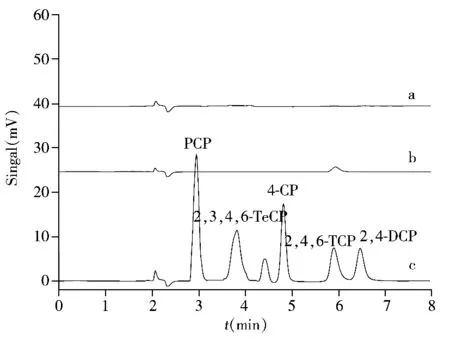
图7 空白模拟海水样品(a)、实际样品(b)以及模拟海水样品加标后(加标浓度2 μg/L)(c)的HPLC图谱Fig.7 HPLC chromatograms of the blank synthesized sea water(a),real sea water sample(b)and spiked sample(2 μg/L)(c)
[1]Chao Y Y,Tu Y M,Jian Z X,Wang H W,Huang Y L.J.Chromatogr.A,2013,1271(1):41-49.
[2]Yi R,Li L C,Wang X,Gao J,Yan F.Environ.Monitor.China(易睿,李利聪,汪霄,高娟,颜峰.中国环境监测),2012,28(3):90-93.
[3]Pavon J L P,Ferreir A M C,Laespada M E F,Cordero B M.J.Chromatogr.A,2009,1216(7):1192-1199.
[4]Huang H L,He Z G,Li M,Mo Z F,Shan X W,Liang Y.Chin.J.Anal.Chem.(黄慧玲,何志贵,李渺,莫振飞,单锡文,梁毅.分析化学),2007,35(9):1369-1372.
[5]Ding Z Q,Zhang Q Y.Chin.J.Anal.Chem.(丁宗庆,张琼瑶.分析化学),2010,38(10):1400-1404.
[6]Ki M D,Han J,Cho I Y.Anal.Bioanal.Chem.,2013,405(1):377 -387.
[7]Zhou C H,Tong S S,Chang Y X,Jia X,Zhou W H.Electrophoresis,2012,33(8):1331 -1338.
[8]Almeda S,Nozal L,Arce L,Valcarcel M.Anal.Chim.Acta,2007,587(1):97-103.
[9]Fu S L,Ding L,Dai H,Zhu S H,Jiao Y N,Gong Q,Chen J T,Wu X H,Wang L B.J.Instrum.Anal.(付善良,丁利,戴华,朱绍华,焦艳娜,龚强,陈继涛,吴新华,王利兵.分析测试学报),2011,30(8):847-852.
[10]Zhou A M,Wang Z J,Chen J L,Shen H Y,Hu M Q,Dong X Y,Xia Q H.J.Instrum.Anal.(周阿蒙,王哲君,陈君良,沈昊宇,胡美琴,董新艳,夏清华.分析测试学报),2014,33(11):1219-1223.
[11]Peng L,Wang Y Z,Zeng H,Yuan Y.Analyst,2011,136:756 -763.
[12]Oriol B,Montserrat L,Cristina P,Cristina P,Franck L D,Florence P K.Anal.Methods,2013,5:6297-6305.
[13]Zhang Z B,Chen Z D,Liu L S,Wang Z D.Principles and Applications of Marine Chemistry:Chemical Chinese Offshore Marine.Beijing:Marine Press(张正斌,陈振东,刘莲生,王肇鼎.海洋化学原理和应用:中国近海的海洋化学.北京:海洋出版社),1999.
[14]Zhao Y G,Chen X H,Pan S D,Zhu H,Shen H Y,Jin M C.J.Mater.Chem.A,2013,1:11648-11658.
[15]Ozkutuk E B,Ersoz A,Denizli A,Say R.J.Hazard.Mater.,2008,157:130 -136.
[16]Pena-Pereira F,Cabaleiro N,Calle I D L,Costas M,Gil S,Lavilla I,Bendicho C.Talanta,2011,85:1100-1104.
[17]Bagheri H,Mir A,Babanezhad E.Anal.Chim.Acta,2005,532:89-95.
[18]Santana C M,Padrón M E T,Ferrera Z S,Rodríguez J J S.J.Chromatogr.A,2007,1140:13 -20.
[19]Fiamegos Y C,Kefala A P,Stalikas C D.J.Chromatogr.A,2008,1190:44-51.
[20]López-Jiménez F J,Rubio S,Pérez-Bendito D.J.Chromatogr.A,2008,1195:25 -33.
[21]Guo L,Lee H K.J.Chromatogr.A,2011,1218:4299-4306.
[22]Marchese S,Gentili A,Perret D,Sergi M,Notari S.Chromatographia,2004,59:411-417.
[23]Liu X,Yin J,Zhu L,Zhao G,Zhang H.Talanta,2011,85:2451-2457.
[24]Yang F,Shen R,Long Y,Sun X,Tang F,Cai Q,Yao S.J.Environ.Monit.,2011,13:440 -445.
[25]Li J,Zhao X,Shi Y,Cai Y,Mou S,Jiang G.J.Chromatogr.A,2008,1180:24-31.
[26]Pan S D,Zhou L X,Zhao Y G,Chen X H,Shen H Y,Cai M Q,Jin M C.J.Chromatogr.A,2014,1362:34-42.
猜你喜欢
现代畜牧科技(2021年9期)2021-10-13
趣味(数学)(2019年12期)2019-04-13
儿童故事画报·发现号趣味百科(2017年10期)2018-03-13
军事文摘·科学少年(2017年4期)2017-06-20
作文周刊·小学一年级版(2016年39期)2017-03-03
材料科学与工程学报(2016年2期)2017-01-15
应用化工(2014年8期)2014-08-08
江西理工大学学报(2013年1期)2013-03-20
郑州大学学报(理学版)(2013年3期)2013-03-11
儿童时代·幸福宝宝(2009年2期)2009-02-19
