
氧瓶燃烧-离子色谱电导检测法测定格列喹酮主环物中水合氯醛的含量
2015-01-01 02:34:44王东武刘志娟侯倩倩安冬华
分析测试学报 2015年10期
王东武,刘志娟,许 峰,刘 斐,侯倩倩,安冬华
(1.迪沙药业集团 国家认定企业技术中心质量研究院,山东 威海 264200;2.中国兵器工业集团 第五三研究所,山东 济南 250031;3.迪沙药业集团 迪素质管部,山东 威海 264200)
格列喹酮属于第二代磺脲类降糖药,具有高活性胰岛β细胞亲和作用,能够与胰岛β细胞膜上的特异性受体结合,诱导产生适量胰岛素以降低血糖浓度。该药品安全有效,毒副作用少,对于新发的二型糖尿病有显著疗效,尤适用于糖尿病伴有肾损伤或肾移植的病人[1-4]。格列喹酮按照化学药品注册分类属于6类化药,其工业化大生产的工艺路径是:格列喹酮主环物与对乙胺苯磺酰胺缩合后,再与N-环己基甲酰胺进行脱氢缩合生成格列喹酮(见图1)。格列喹酮主环物作为合成格列喹酮的关键起始物料,按照国家食品药品监督管理局药审中心颁布的《化学药物杂质研究的技术指导原则》要求,需对格列喹酮主环物进行杂质研究。由格列喹酮主环物生产工艺可知,合成格列喹酮主环物过程中使用了大量的水合氯醛,由于水合氯醛具有一定的毒性,大剂量可引起昏迷、麻醉、肝损伤、呼吸抑制及心血管功能改变,甚至死亡,因此需要建立严格的质量标准以控制格列喹酮主环物中水合氯醛的含量[5-9]。
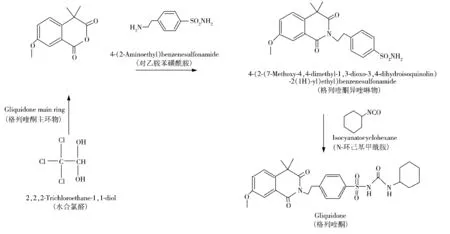
图1 格列喹酮的合成工艺路线图Fig.1 Chart of gliquidone synthesis process
目前水合氯醛含量的测定方法主要为紫外分光光度法和气相色谱法[10-12],其中分光光度法易受格列喹酮主环物的干扰,而气相色谱法检测时,由于水合氯醛结构中碳氢键数目较少,燃烧时产生的碳氢自由基少,导致灵敏度低,难以满足准确检测格列喹酮主环物中痕量水合氯醛的要求。
氧瓶燃烧法作为一种成熟的样品前处理方法已广泛应用于有机溴、氯、磷及硫等元素的含量测定[13-15],该方法使有机溴、氯、磷及硫等元素通过富氧燃烧转化为无机元素,然后通过滴定、离子色谱等方法测定无机元素的含量间接测定这些有机元素。离子色谱法在离子检测方面具有灵敏度高、专属性好等优点,特别是近年来离子色谱填料技术、电化学检测技术及离子抑制技术的快速发展,使得离子色谱法在分析检测领域得到了普遍应用[16-21]。以氧瓶燃烧法进行样品处理,离子色谱法进行检测的分析方法,已广泛应用于化工、生物、医药等样品中有机硫、氯等元素的含量测定[22-25]。
1 实验部分
1.1 仪器与试剂
Toson IC-2010离子色谱仪(日本Toson公司),配有高压二元梯度泵、自动进样器、柱温箱、自动交换凝胶离子抑制系统、电导检测器及IC-2010数据工作站;色谱柱:日本TSKgec SuperIC-Anion HS阴离子色谱柱(4.6 mm i.d.×10 cm,3.5 μm);TSK suppress IC-A阴离子抑制胶(日本Toson公司);热电超纯水系统EASY pureⅡ(美国热电公司);滤纸(杭州特种纸业有限公司)。
Cl-,F-,I-,SO2-4,NO-2,NO-3,PO3-4,Br-对照品溶液,浓度均为1 000 mg/L(国防科技工业应用化学一级计量站);NaOH(成都科龙化工试剂厂,批号20120416)、无水NaCO3(天津市鼎盛鑫化工有限公司,批号20141015)、NaHCO3(天津市巴斯夫化工有限公司,批号20081103)、水合氯醛(成都市科龙化工试剂厂,批号20110411)均为分析纯;格列喹酮主环物(迪沙药业集团原料药研究二院,批号分别为:141205M1,141205M2,141205M3)。
1.2 溶液制备
供试品溶液:准确称取格列喹酮主环物约10 mg,包裹于滤纸中,放入铂丝钳口,用酒精灯引燃,迅速置于盛有20 mL 0.05 mol/L NaOH的燃烧瓶中,瓶中富氧,燃尽后,以超纯水封口,待吸收1 h后,转入100 mL容量瓶中,烧瓶多次冲洗,洗液均转入容量瓶中,与吸收液合并,超纯水定容至刻度后,摇匀。
供试品溶液加标:将上述洗液与吸收液合并后,加入一定体积的Cl-标准溶液,超纯水定容至刻度,摇匀即得。
滤纸对照溶液:取滤纸(大小同包裹样品用滤纸),放入铂丝钳口,用酒精灯引燃,迅速置于盛有20 mL 0.05 mol/L NaOH的燃烧瓶中,进行富氧燃烧,燃尽后,超纯水封口,待吸收1 h后,转入100 mL容量瓶中,烧瓶多次冲洗,洗液均转移至容量瓶中,与吸收液合并,超纯水定容至刻度后,摇匀。
Cl-对照品储备液:精密移取1 000 mg/L的Cl-标准溶液1 mL至100 mL容量瓶中,超纯水稀释后定容至刻度,摇匀即得。
Cl-对照溶液:精密量取Cl-对照品储备液0.5 mL至100 mL容量瓶中,超纯水稀释后定容至刻度,摇匀即得。
干扰离子对照溶液:分别精密量取浓度均为1 000 mg/mL的F-,Cl-,NO-3,I-,PO3-4,SO2-4,Br-,NO-2标准溶液各1 mL至100 mL容量瓶中,加超纯水定容至刻度,摇匀,精密量取该溶液1 mL至25 mL容量瓶中,加超纯水稀释至刻度,摇匀即得。
本研究证实注射用雷贝拉唑钠不仅对急性溃疡,如醋酸性模型和幽门结扎法模型有效,还对消炎痛法模型等慢性溃疡有效,同时还对大鼠反流性食管炎以及半胱胺型十二指肠溃疡均具有良好的抑制作用,与临床适用症一致。

方法开发溶液的配制:精密称取格列喹酮主环物约10 mg,用滤纸包裹,置于铂丝钳口,夹紧,引燃,迅速插入盛有20 mL 0.05 mol/L NaOH的富氧烧瓶中,燃尽后,超纯水封口。待吸收1 h后,吸收液转入100 mL容量瓶中,烧瓶多次冲洗,合并洗液与吸收液,加入0.4 mL 10 mg/L的Cl-,超纯水定容后,摇匀即得。此时溶液中氯离子的浓度约为0.056 mg/mL。
加标供试品:精密称取格列喹酮主环物约20 g加至50 mL二氯甲烷中,加入3种质量(12.48,15.60,18.72 mg)的水合氯醛,搅拌至全溶,室温真空抽干,得样品。精密称取该样品约10 mg,用滤纸包裹,置于铂丝钳口,夹紧,引燃后,迅速插入盛有20 mL 0.05 mol/L NaOH水溶液中的500 mL富氧烧瓶中,密闭烧瓶后,倒置烧瓶待样品炽灼烧尽后,超纯水封口。1 h后,吸收液转入100 mL容量瓶中,烧瓶多次冲洗,合并洗液与吸收液,超纯水定容,摇匀后即得加标80%供试品,加标100%供试品,加标120%供试品。
1.3 分析方法
Toson IC-2010离子色谱仪;色谱柱为TSKgec SuperIC-Anion HS阴离子色谱柱(4.6 mm i.d.×10 cm,3.5 μm);流动相为7.5 mmol/L NaHCO3-1.1 mmol/L Na2CO3;流速为1.0 mL/min;柱温40℃;进样量为30 μL;检测器为电导检测器。
2 结果与讨论
2.1 滤纸选择
通过对比研究杭州特种纸业有限公司和抚顺市民政滤纸厂两厂家滤纸,发现杭州特种纸业有限公司滤纸中的Cl-含量明显低于后者,故实验均选用杭州特种纸业有限公司所生产的滤纸。
2.2 色谱条件的优化
阴离子色谱法中常用的流动相有NaOH溶液和NaHCO3-Na2CO3溶液,分别选择2.5 mmol/L NaOH溶液、5.0 mmol/L NaOH溶液、7.5 mmol/L NaHCO3-1.1 mmol/L Na2CO3溶液为流动相,按照“1.3”方法,进样方法开发溶液。结果显示,以氢氧化钠溶液作流动相时,基线波动较大,且样品峰拖尾严重,峰形对称性差;以NaHCO3-Na2CO3溶液为流动相时基线平缓,峰形对称性较好,故选择NaHCO3-Na2CO3溶液作为流动相。通过对NaHCO3-Na2CO3溶液浓度的考察,最终选择7.5 mmol/L NaHCO3-1.1 mmol/L Na2CO3水溶液为流动相。
以7.5 mmol/L NaHCO3-1.1 mmol/L Na2CO3水溶液为流动相,按照“1.3”方法,分别在柱温25,30,40℃条件下进样方法开发溶液。结果显示柱温越高,色谱峰展宽越小,峰形越好,分析时间越短,考虑到TSKgec SuperIC-Anion HS阴离子柱允许使用的最高柱温为40℃,因此实验选择柱温为40℃。以7.5 mmol/L NaHCO3-1.1 mmol/L Na2CO3水溶液为流动相,按照“1.3”方法,分别在流速为0.8,1.0,1.2 mL/min条件下进样方法开发溶液,结果显示,流速越高,色谱峰展宽越小,峰形对称性更佳,分析时间更短,考虑到阴离子柱的耐压情况及保养维护,实验选择最佳流速为1.0 mL/min。
2.3 供试样品制备原理
采取氧瓶燃烧法使水合氯醛中的有机氯转化为无机氯,经NaOH溶液吸收后以Cl-形式存在。选择体积为500 mL的氧瓶以获取充足的氧气容量使样品燃烧充分,确保有机氯能够完全转化为无机氯。吸收液为20 mL 0.05 mol/L NaOH,相对样品中转化的痕量Cl-,该吸收液NaOH足量,以确保能够完全吸收Cl-。
由于滤纸中不可避免会引入Cl-,因此在供试品制备时,要平行制备滤纸对照溶液,计算供试品中Cl-含量时予以扣除。
2.4 分析方法的验证
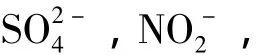
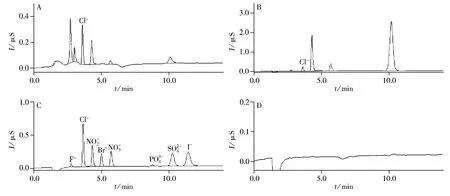
图2 方法专属性实验色谱图Fig.2 Chromatograms of specificity of the method
2.4.2 线性及范围、检出限与定量下限 分别精密量取Cl-储备液0.25,0.4,0.5,0.6,0.8 mL至5个100 mL容量瓶中,加超纯水定容至刻度,摇匀,配成浓度为0.025,0.04,0.05,0.06,0.08 mg/L的线性溶液。按照“1.3”方法进样分析,以Cl-峰面积(y)对Cl-浓度(x,mg/L)进行线性回归,结果显示,Cl-浓度在0.025~0.080 mg/L范围线性关系良好,能够满足Cl-含量的测定要求,回归方程为y=15.29x-0.1316,r=0.999 9。
精密量取10 mg/L的Cl-储备液进行逐级稀释,按照“1.3”方法进样分析,以仪器信噪比(S/N)为3计算方法的检出限,得到Cl-的检出限为0.01 μg/L。以S/N约为10时计算方法的定量下限,得到Cl-的定量下限为0.05 μg/L,相当于水合氯醛定量下限为0.078 μg/L。
2.4.3 重现性 按照“1.3”方法进样分析Cl-标准对照溶液,连续进样6针,计算Cl-色谱峰面积的RSD为1.7%,表明方法的精密度良好。按照“1.3”方法,由不同的实验人员,在不同的实验日期,进样分析Cl-标准对照溶液,分别连续进样6针,计算Cl-色谱峰面积的RSD为0.50%,12针样品的RSD为1.4%,表明方法的中间精密度良好。
2.4.4 准确度 按照“1.3”方法,对加标80%供试品,加标100%供试品,加标120%供试品分别进行测定,扣除样品中由滤纸引入的Cl-量,以外标法计算加标回收率,得到Cl-的加标回收率为92.9%~103.4%,RSD为6.4%(表1),结果表明该方法的准确度良好。
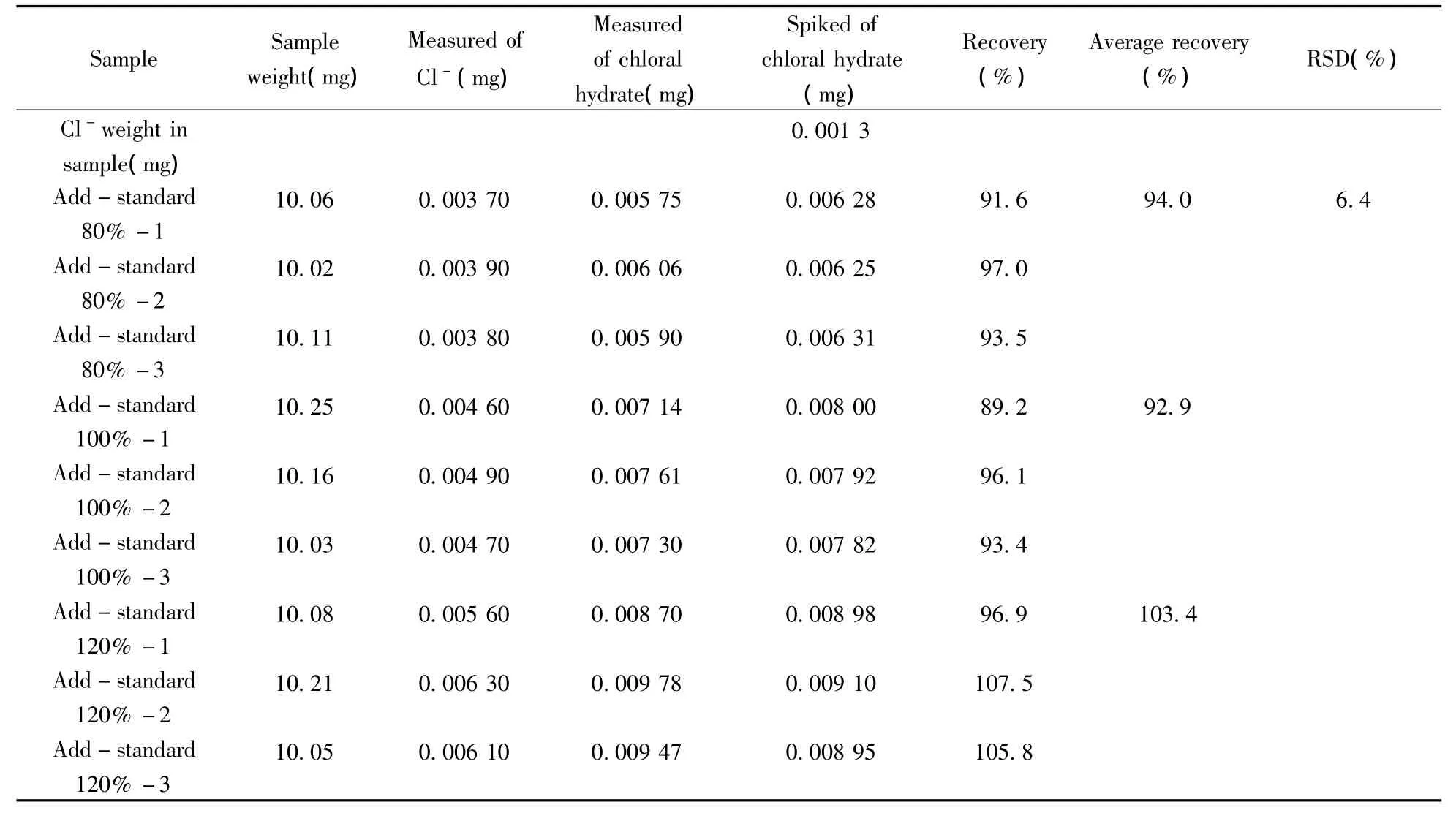
表1 回收率实验结果Table 1 Result of recovery test
2.4.5 样品稳定性 按“1.2”供试品溶液加标的配制方法,加入0.4 mL 10 mg/mL的Cl-,用水定容后,摇匀即得测试样品。按照“1.3”方法,分别在0,1,2,4,8,12 h对测试液进行测定,结果显示Cl-峰面积的RSD为2.8%,表明样品在12 h内的稳定性良好。
2.4.6 方法耐用性 按“1.2”供试品溶液加标的配制方法,加入0.5 mL 10 mg/mL的Cl-,用水定容后,摇匀即得样品稳定性测试溶液。按照“1.3”方法,分别设置流速为0.8,1.0,1.2 mL/min;柱温为38,40,42℃;缓冲液浓度为8.25 mmol/L NaHCO3-1.21 mmol/L Na2CO3、7.5 mmol/L NaHCO3-1.1 mmol/L Na2CO3、6.75 mmol/L NaHCO3-0.99 mmol/L Na2CO3;考察流速、柱温以及盐浓度变化时对Cl-含量测定结果的影响。结果显示:柱温、流速、缓冲液浓度在一定范围内变化对样品中氯离子的分离和含量测定无明显影响,均能达到基线分离;Cl-含量测定结果的RSD分别为5.9%,2.7%,5.0%(表2),表明该方法的流速耐用性、柱温耐用性以及盐浓度耐用性均良好。
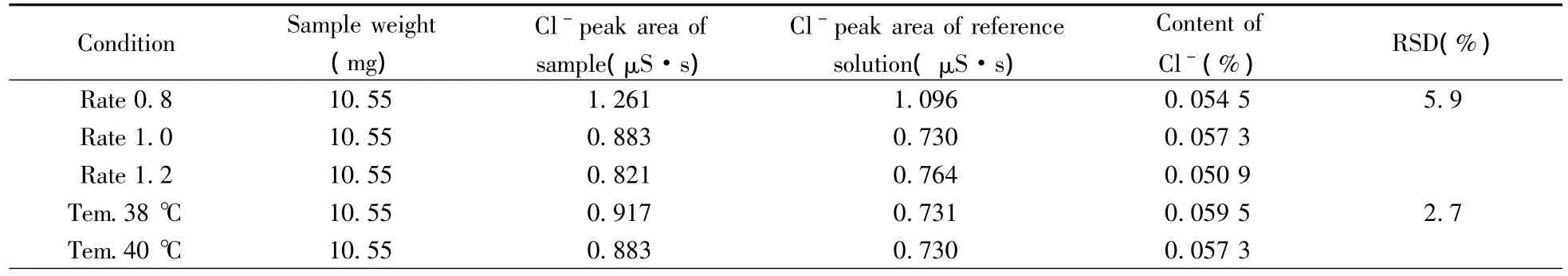
表2 耐用性实验结果Table 2 Result of robustness test
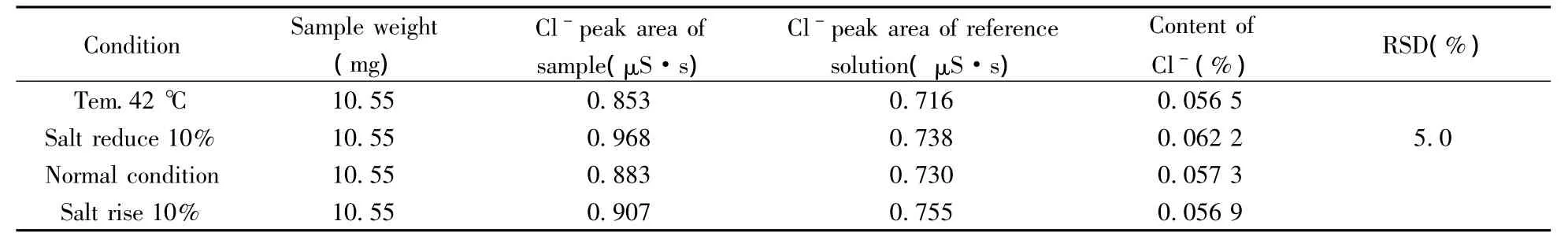
(续表2)
2.4.7 样品检测 按照“1.3”方法,分别进样分析3批供试品溶液,记录色谱图,以外标法计算Cl-含量。结果显示,3批样品中的Cl-含量均低于0.02%,折换成水合氯醛,3批样品中的水合氯醛含量均不大于0.03%,符合质量标准中对格列喹酮主环物中水合氯醛含量的要求。
3 结论
该方法通过氧瓶燃烧法使格列喹酮主环物中的有机氯转化为无机氯离子,经氢氧化钠水溶液吸收后,运用离子色谱法测定供试品溶液中的氯离子含量,从而间接测定格列喹酮主环物中水合氯醛的含量。该方法准确、有效、灵敏,能够满足格列喹酮主环物中水合氯醛的定量检测要求。
[1]Malaisse W J.Drugs R D.,2006,7(6):331 -337.
[2]Türk T R,Bandur S,Nürnberger J,Kribben A,Mann K,Philipp T,Heemann U,Viklicky O,Witzke O.Clin.Nephrol.,2008,1(70):26-32.
[3]Ke J T,Li M,Xu S Q,Zhang W J,Jiang Y W,Cheng L Y,Chen L,Lou J N,Wu W.J.Endocrinol.,2014,220(2):129-141.
[4]Ke J T,Li M,Jiang Y W,Xu S Q,Zhang W J,Wu W,Lou J N.Chin.J.Hospital Pharm.(柯剑婷,李宓,姜永玮,许世清,张文健,伍卫,娄晋宁.中国医院药学杂志),2013,10:789-792.
[5]Lu Y H.Herald of Medicine(芦映红.医药导报),2011,1:127-128.
[6]Jiang X,Gao L,Zhang Y,Wang G,Liu Y,Yan C,Sun H.J.Cardiovasc.Med.(Hagerstown),2011,12(10):732-735.
[7]Han P,Song H,Yang P,Xie H,Kang Y J.Cardiovasc.Toxicol.2011,11(2):128 -133.
[8]Zhang J,Grindstaff R D,Thai S F,Murray S A,Kohan M,Blackman C F.Cell Biol.Toxicol.,2011,27(3):207 -216.
[9]Lock E A,Reed C J,McMillan J M,Oatis J E Jr,Schnellmann R G.Toxicology,2007,230(2/3):234 -243.
[10]Deng Y H,Ye L J,Liang Y,Dai L F.Chin.J.Spectrosc.Lab.(邓岩辉,叶丽杰,梁岩,戴莉菲.光谱实验室),2011,5:2299-2301.
[11]Sun W,Lu H T,Zhao X M.China Chlor-Alkali(孙伟,鲁洪涛,赵雪梅.中国氯碱),2003,7:44.
[12]Tang H,Meng X J,Wang F Q.China Medical Herald(唐虹,孟祥军,王凤秋.中国医药导报),2012,20:109-110.
[13]Wang T L,Yuan X Y,Guan T T,Liu Y J,Yi S J,Tang C R,Zeng Y L.Chin.J.Anal.Lab.(王天伦,元晓云,关婷婷,刘亚娟,易守军,唐春然,曾云龙.分析试验室),2015,2:143-145.
[14]Moraes D P,Pereira J S,Diehl L O,Mesko M F,Dressler V L,Paniz J N,Knapp G,Flores E M.Anal.Bioanal.Chem.,2010,397(2):563-570.
[15]Geng W H,Furuzono T,Nakajima T,Takanashi H,Ohki A.J.Hazard Mater.,2010,176(1/3):356 -360.
[16]Yang L,Hou Y,Wang B X,Yang S H,Zou Y,Yang Y,Yang Y,Liu J.J.Instrum.Anal.(杨蕾,侯英,王保兴,杨式华,邹悦,杨燕,杨勇,刘静.分析测试学报),2010,29(2):165-170.
[17]Ma Y J,Zhang X,Yu H.J.Instrum.Anal.(马亚杰,张欣,于泓.分析测试学报),2012,31(12):1591-1594.
[18]Zeng X L,Ye M L,Chen Y X,Zhu Y.J.Instrum.Anal.(曾雪灵,叶明立,陈永欣,朱岩.分析测试学报),2006,25(3):92-94.
[19]Zhang S F,Lin Z Z,Chen X Z,Zhang D L,Wang L S.J.Instrum.Anal.(张水锋,林珍珍,陈小珍,张东雷,王立世.分析测试学报),2012,31(1):85-89.
[20]Chen Q,Yu H,Meng L M,Huang X.J.Instrum.Anal.(陈倩,于泓,孟令敏,黄旭.分析测试学报),2011,30(1):38-42.
[21]Li Y,Liu H.J.Instrum.Anal.(李月,刘晃.分析测试学报),2012,31(1):66-70.
[22]Zhang S,Zhao T,Wang J,Qu X L,Chen W,Han Y.J.Chromatogr.Sci.,2013,51:65 -69.
[23]Jiang J H.Chin.J.Chromatogr.(江锦花.色谱),2006,24(4):423.
[24]Lian J,Liu Z X,Wen L Q,Xie H X.Hebei Journal of Forestry and Orchard Research(连军,刘志轩,文丽青,解慧欣.河北林果研究),2003,18(Suppl):282-284.
[25]Murayama M,Suzuki M,Takitani S.J.Chromatogr.,1989,463:147-152.
猜你喜欢
健康之家(2022年9期)2022-05-30 08:20:30
中南民族大学学报(自然科学版)(2022年3期)2022-05-08 03:51:04
节能与环保(2022年3期)2022-04-26 14:32:44
化工科技(2020年1期)2020-03-16 08:41:06
国外医药(抗生素分册)(2015年3期)2015-07-12 12:28:27
中国当代医药(2015年33期)2015-03-01 02:09:18
当代临床医刊(2015年3期)2015-01-21 01:05:50
中国有色冶金(2014年6期)2014-08-10 12:29:04
中国药业(2014年19期)2014-05-17 03:12:00
上海计量测试(2012年2期)2012-04-14 09:58:34
