
皂苷类似物与肾素的分子对接和结合能分析
2014-12-25 02:28张海玲光翠娥汪俊卿桑尚源
食品与生物技术学报 2014年10期
张海玲, 光翠娥*, 江 波, 汪俊卿, 桑尚源
(1.食品科学与技术国家重点实验室 江南大学,江苏 无锡 214122;2.江南大学 生物工程学院,江苏 无锡214122)
随着现代社会人口老龄化进程的不断加快,罹患高血压的人群逐年增加,心血管疾病、糖尿病、充血性心脏衰竭、肾功能异常等并发症同时增加。肾素-血管紧张素系统(RAS)参与体液和血压的内分泌调控进而控制血管收缩,调节心血管功能和肾功能并维持身体电解质平衡,RAS过度激活导致血管结构和功能损伤引起高血压疾病[1]。肾素是催化RAS的关键限速酶,通过抑制肾素的活性则可阻断RAS,因此肾素一直是被人们关注的重要靶点。很多研究发现,一些皂苷类的化合物对心脑血管疾病以及肾疾病有良好的治疗效果,已经确定的大豆皂苷I及其类似物大豆皂苷II、甘草皂苷、单葡萄糖醛酸甘草皂苷元与肾素具有生物活性作用。
随着核磁共振和X射线衍射方法的发展,越来越多的蛋白质的三维结构被测出来。分子对接是分子模拟的方法之一,研究受体大分子和配体小分子结合作用,预测分子之间最佳的结合模式,并通过研究两者对接结合的能量、作用位点、关键残基等方面评价受体与配体的相互作用[2],是预测和研究小分子配体与大分子蛋白质受体相互作用模式的重要方法[3]。因此,在基于肾素和皂苷结构的基础上进行分子对接,进而分析对接复合物相互作用的轮廓是否存在相似性,并揭示肾素的活性与皂苷相互作用氢键、疏水作用和重要残基氨基酸,探讨皂苷对于肾素的抑制机理。
1 材料与方法
1.1 肾素的结构与模型的选取
从蛋白质晶体结构数据库(http://www.rcsb.org/pdb/)中下载初始结构为Renin-indole-piperazinj复合 物 结 构 (PDB ID:3OOT), 小分子配体为C8H15NO6和 C28H28FN3O2,利用 Discovery Studio 2.5(DS)软件去掉小分子配体和水分子得到肾素的三维结构并作为受体用于后续的对接过程。
1.2 大豆皂苷I及其类似物结构的获取
在 chemicalbook网站库 (http://www.chemicalbook.com)中下载大豆皂苷I三维结构,然后在大豆皂苷I的三维结构基础上利用pymol软件[4](http://pymol.org)绘制出大豆皂苷 II(soyasaponin II)、甘草皂苷 (glycyrrhizin)、单葡萄糖醛酸甘草皂苷元(MGGM)的三维结构,图1为4种皂苷的分子结构。

图1 4种皂苷分子结构Fig.1 Molecular structure of four saponins
1.3 肾素与皂苷的分子对接
分子对接使用的是北京创腾科技有限公司的Discovery Studio(DS)软件,肾素作为为受体并删掉其中的水分子,4种皂苷作为对接配体,将距肾素活性中心的Asp226 20×10-9cm内的范围定义为拟对接区域,并添加CHARMm力场,按照王红寅等[5]人所述方法运用Discovery Studio 2.5软件对配体和受体空间结构进行分子对接,可能构象模式采用Libdock模块进行收集。运用DS程序包对对接复合物的空间结构分别进行两次能量优化,即先采用最陡下降法(Steepest Descent)优化 1 000步,再采用共轭梯度法(Conjugate Gradient)优化2 000步。基于优化后的空间结构使用DS软件统计肾素活性位点内的氨基酸残基,并用Ligplot+软件[6]得到分子间相互作用网,统计受体中与配体产生氢键作用以及疏水作用的氨基酸残基,并对复合物的空间作用力进行分析。
1.4 对接复合物结合自由能的计算
分子力学泊松-波尔兹曼表面积(Molecular mechanics Poisson-Boltzmann surface area,MMPBSA)法是一种常用于受体与配体分子对接后计算结合自由能值(binding free energy,ΔGbind)的方法[7-9],ΔGbind值越低说明受体与配体之间的亲和力越高[10,11]。通过MM-PBSA方法可以将分子作用力进行分解,更为直观的分析各种相互作用的贡献。
2 结果与分析讨论
2.1 肾素和抑制剂分子对接和结合自由能计算
4种皂苷和肾素进行分子对接和优化后,按照公式 (1)进行对对接复合物的结合自由能进行计算,结果如表1所示。大豆皂苷I、大豆皂苷II、甘草皂苷以及单葡萄糖醛酸甘草皂苷元与肾素的结合自由能均小于零,表明这些皂苷均能对肾素起到不同程度的抑制作用,其中大豆皂苷I、大豆皂苷II和甘草皂苷对肾素的抑制效果接近,结合自由能均在-35×4.8=-168 kJ/mol左右,而甘草皂苷的抑制效果则相对较差,结合自由能仅为-12.24 kJ/mol。结合自由能反映了受体和配体结合的稳定性[12],将计算所得的结合自由能与4种皂苷的IC50值进行比较,发现使用MM-PBSA计算所得的结果能够较好的契合Saori Takahashi[13]等人的实验结果,如图2所示,R2为0.79,表明使用该方法预测皂苷对肾素的抑制能力具有一定的可行性。

表1 对接复合物的结合自由能Table 1 Binding free energy of docking compounds
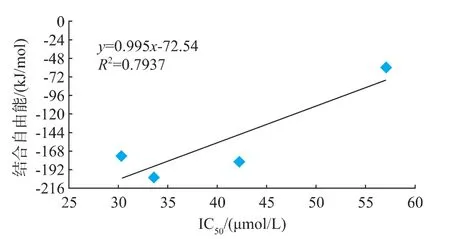
图2 结合自由能和IC50值相关性Fig.2 Correlation between binding free energy and IC50
表1中列出了对结合自由能贡献的各项能量值,通过图3中各种作用力可以看出,ΔEvdW,ΔEelec和ΔGSUR均为负值,表明4种肾素抑制剂和肾素之间的范德华力,静电相互作用和非极性溶剂化能对于肾素抑制剂和肾素的结合自由能都起到促进作用。在4种皂苷和肾素形成的复合物中,ΔEvdW和ΔEelec在ΔGbind中占有主导地位,表明范德华力和静电相互作用对于4种皂苷和肾素之间的结合能作用贡献比较大(>90%)。
大豆皂苷I、大豆皂苷II、甘草皂苷和单葡萄糖醛酸甘草皂苷元与肾素形成复合物的范德华力分别为-93.97、-81.43、-72.79、-64.68 kJ/mol,大豆皂苷I和大豆皂苷II的范德华力比甘草皂苷和单葡萄糖醛酸甘草皂苷元大。虽然静电相互作用也对4种抑制剂与肾素复合物的形成有促进作用,但是较强的极性溶剂化能抵消这部分作用力,阻碍皂苷和肾素的结合。
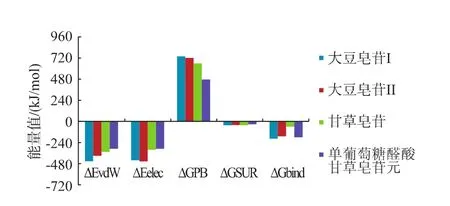
图3 结合自由能各能量示意图Fig.3 Diagram:each energy contribution value in binding free energy of eight groups complexes
2.2 皂苷和肾素形成的疏水作用分析
非极性相互作用能对结合自由能有促进作用,肾素可与4种皂苷结合形成较多的疏水相互作用。表2为通过Ligpiot软件统计肾素和4种皂苷相互作用的疏水氨基酸残基。
通过图4分析可得,氨基酸残基Ala229、Asp38、Asp226、Gly228和 Tyr83与 4种皂苷在肾素与4种皂苷的结合过程中都参与形成疏水相互作用,表明这4种氨基酸是参与肾素和皂苷抑制剂结合的重要氨基酸位点。此外,分析还发现Ala317、Gln19、Met303、Thr18、Tyr20 和 Val36 在肾素与大豆皂苷I、大豆皂苷II和甘草皂苷结合过程中形成疏水相互作用,His301同时参与肾素与大豆皂苷I、大豆皂苷II和单葡萄糖醛酸甘草皂苷元结合形成疏水相互作用,Ser41同时大豆皂苷I、甘草皂苷和单葡萄糖醛酸甘草皂苷元形成疏水作用,Gly40与大豆皂苷II、甘草皂苷和单葡萄糖醛酸甘草皂苷元形成疏水作用。
综上所述,范德华力是肾素与4种皂苷结合形成复合物主要驱动能之一,大豆皂苷I和大豆皂苷II的范德华力与甘草皂苷和单葡萄糖醛酸甘草皂苷元相比要高,表明疏水作用可能是大豆皂苷I和大豆皂苷II对肾素抑制效果较好的重要原因。此外,分 析 还 发 现 Ala229、Asp38、Asp226、Gly228 和Tyr83是肾素与皂苷类抑制剂结合过程中形成疏水作用的重要氨基酸残基。

表2 肾素中与抑制剂疏水作用的氨基酸残基Table 2 Amino acid residues of renin reacted with inhibitors

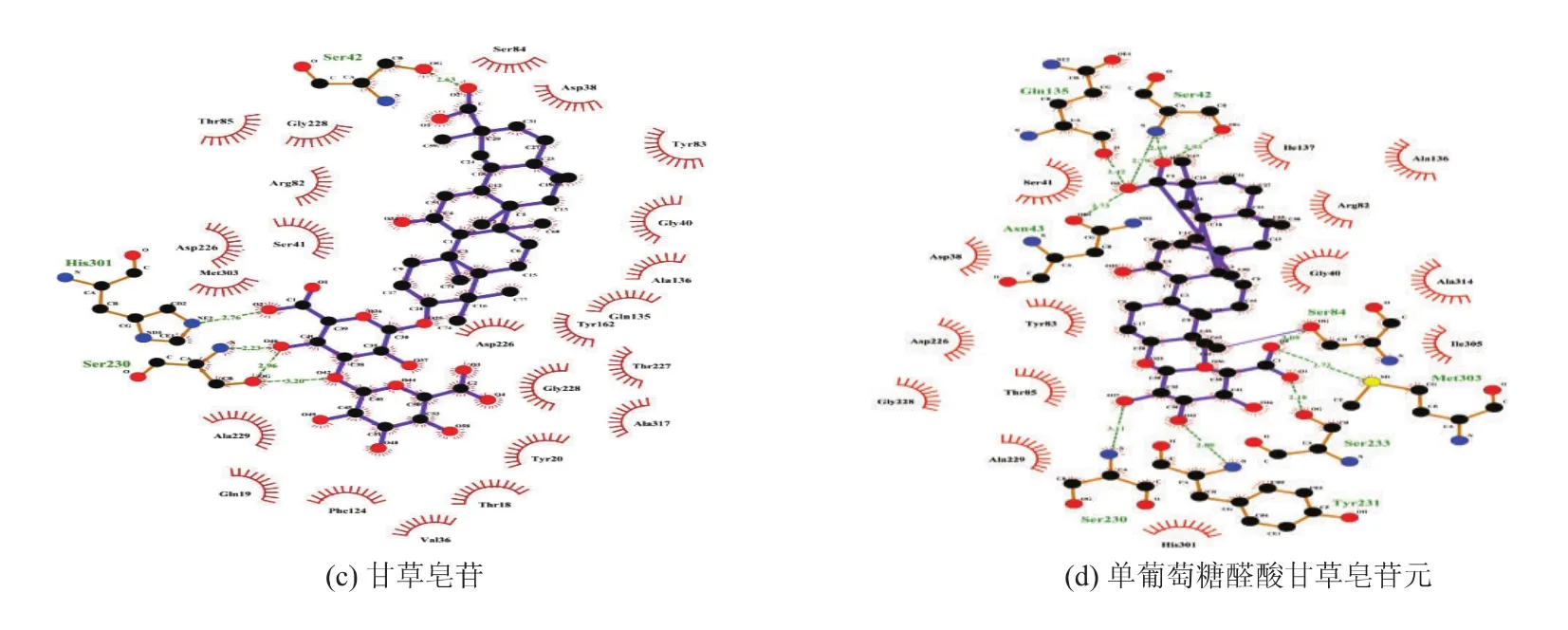
图4 肾素和抑制剂分子间相互作用分析Fig.4 Intermolecular interaction analysis between renin and inhibitors
2.3 皂苷与肾素形成的氢键和配位键分析
通过对4种复合物结合自由能的分析,可以看出极性相互作用很大程度上表现为阻碍肾素和抑制剂皂苷的结合,但是并不是形成极性作用的氨基酸残基都不利于复合物的形成,例如Ser230、Tyr231和Ser84等氨基酸残基在结合过程中都表现出了有利于结合自由能的极性作用,因为在结合过程中它们和皂苷能形成稳定的氢键作用。通过图2中Ligplot软件得到的分子间相互作用网,将体系中较为稳定的氢键作用统计在表3中。虽然一些具有极性作用的氨基酸残基形成了氢键作用,但是远小于溶剂化能作用,结果极性相互作用总体上讲还是阻碍肾素和肾素抑制剂的结合。
2.4 肾素活性口袋的氢键和疏水作用
肾素是肾素原经激活酶转化而生成的一种天冬氨酸蛋白酶,呈二叶体结构,每个叶体上的裂隙内都存在肾素的活性位点,肾素主要的活性位点区域可分为 S3sp,S3,S2,S1,S1’,S2’[14],将 4 种皂苷和肾素进行分子对接后统计肾素活性口袋内参与形成氢键的氨基酸残基,结果如图5和表4所示。
通过表4中肾素活性口袋的氢键分析可得,皂苷和肾素形成的氢键作用主要是在 S3sp、S3、S1、S2、和S2’活性口袋,在肾素的S1’活性口袋内不存在氢键作用。肾素与大豆皂苷I和大豆皂苷II形成氢键的活性口袋十分相似,这是因为大豆皂苷I和大豆皂苷II的结构十分相似,都主要是在S3sp,S3,S2,S1 区域分别和 Tyr162,Ser230,Tyr231 和 Tyr85、Ser84等氨基酸残基形成氢键;甘草皂苷形成氢键的活性口袋则主要位于S3和S2口袋;单葡萄糖醛酸甘草皂苷元形成氢键的活性口袋主要位于S3,S2,S1和S2’区域。通过相同活性口袋内的氨基酸类型分析可得,肾素的氨基酸残基Ser230与4种皂苷都可以形成氢键作用,在S3活性口袋内;Tyr231在S2口袋内可以与大豆皂苷I、大豆皂苷II、单葡萄糖醛酸甘草皂苷元形成氢键作用;活性口袋S1内Ser84在可与大豆皂苷I、大豆皂苷II、单葡萄糖醛酸甘草皂苷元木鳖子皂苷 Ic形成氢键作用。因此,皂苷和肾素形成氢键作用的主要氨基酸残基Ser230、Tyr231 和 Ser84 主要分布在 S3、S1、S2 活性口袋,这些口袋是形成氢键作用是主要活性区域。
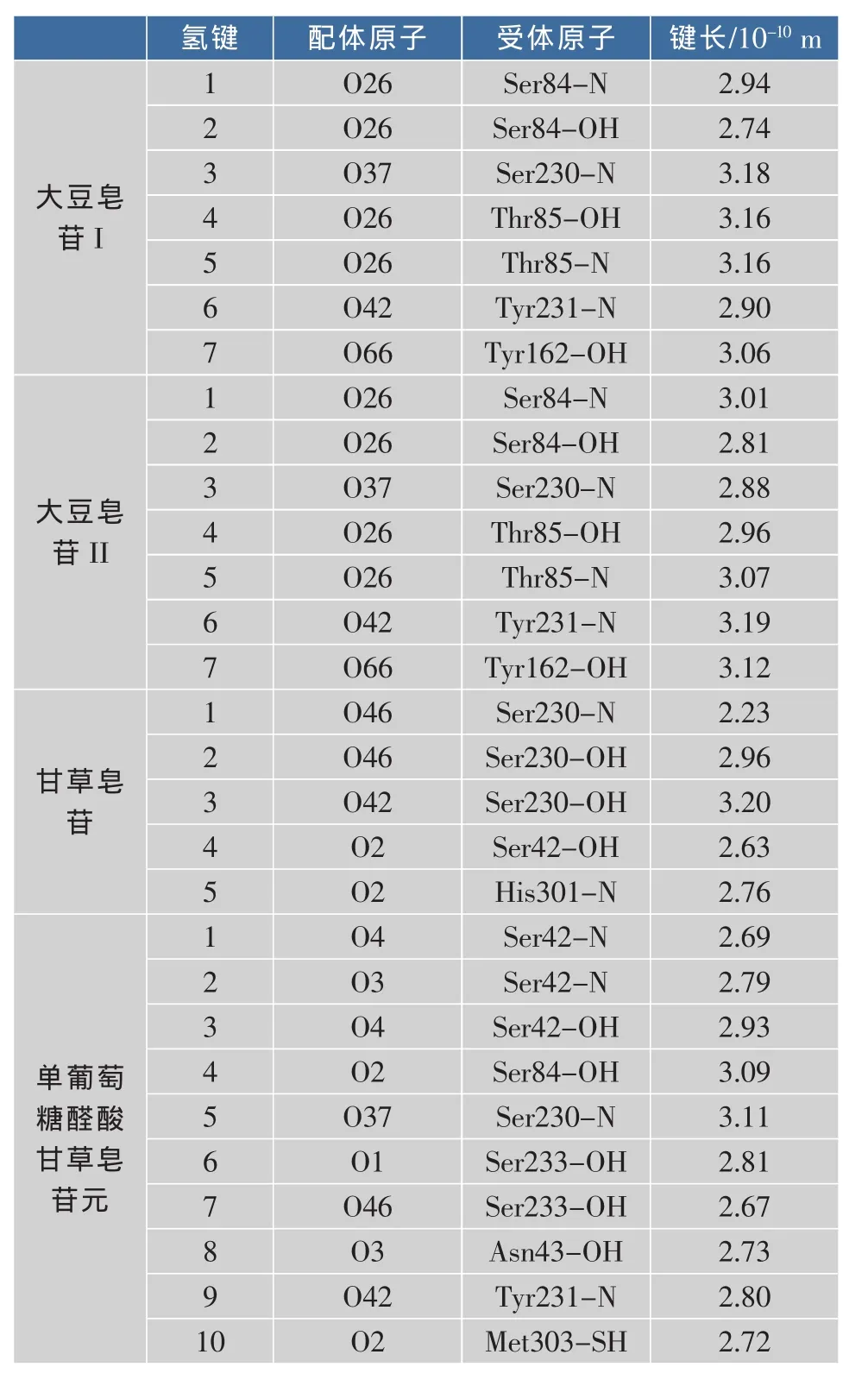
表3 4种复合物中氢键分析Table 3 Hydrogen bond analysis of four compounds
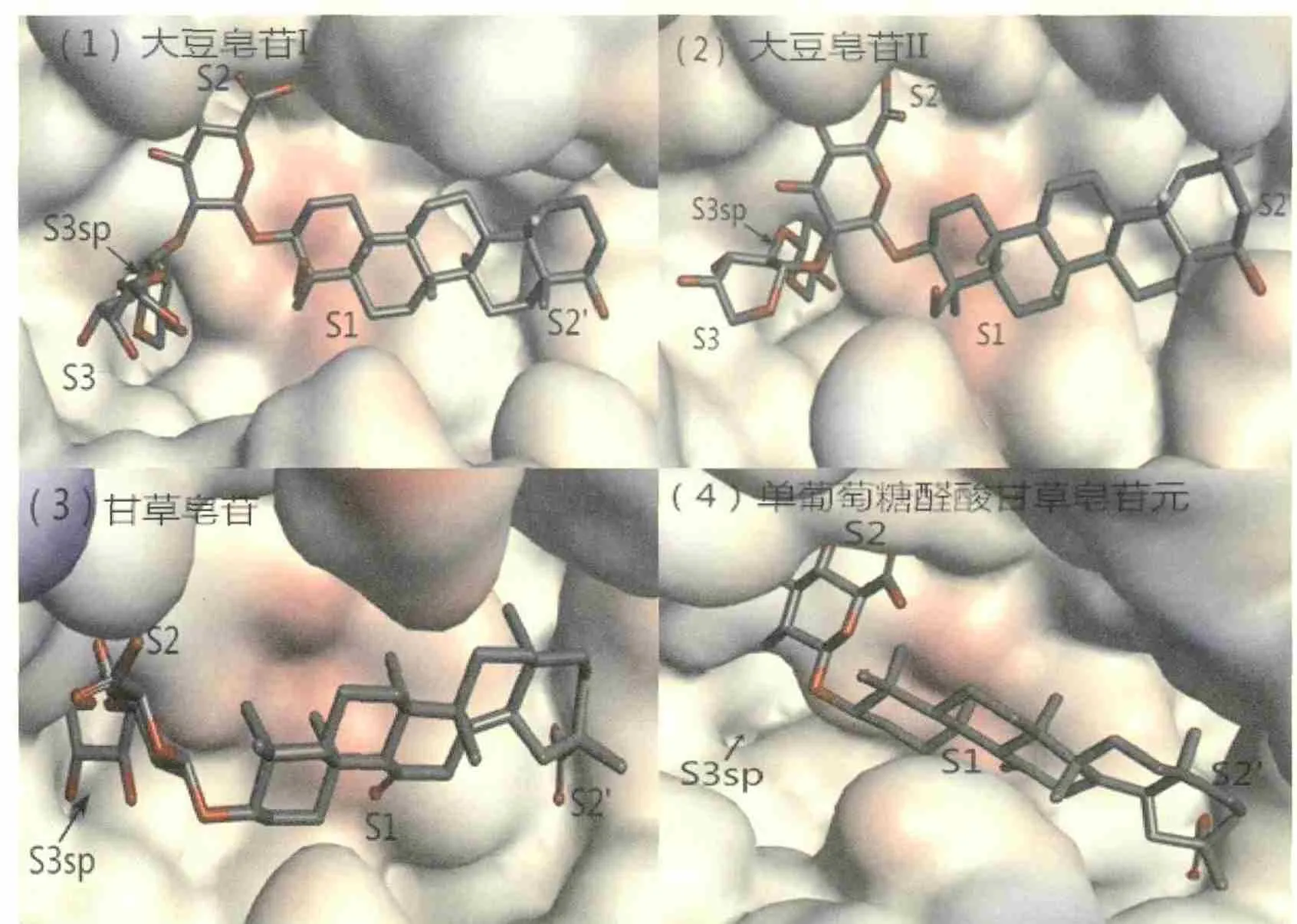
图5 皂苷与肾素的活性口袋的对接Fig.5 Docking between eight saponins and activity pockets of renin
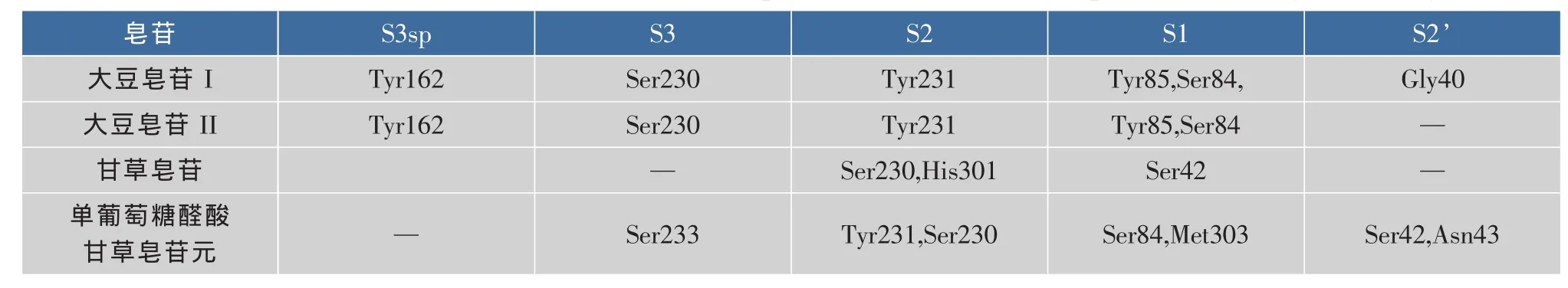
表4 肾素活性口袋内的与皂苷具有氢键作用的氨基酸残基Table 4 Amino acid residues inside renin activity pockets interaction with saponins by hydrogen bondings
将皂苷和肾素之间疏水相互作用的氨基酸残基对应肾素的活性口袋进行分析发现,S3sp,S3,S2,S1,S1’和S2’6个活性口袋内都有较强的疏水相互作用 (见表5)。8组皂苷与氨基酸残基Asp38和Arg82是在S1活性口袋形成疏水作用,Asp226和Tyr83是在S1’活性口袋形成疏水作用,与Ala229在S3活性口袋形成疏水作用;Met303和His301在S2活性口袋和皂苷形成疏水作用;Gly228、Val36和Tyr20在S3sp活性口袋形成与皂苷形成疏水作用;Gln19、Thr18和Thr18在S3活性口袋和皂苷形成疏水作用;Gly40、Gln135、Ser41 和 Ile137 在 S2’活性口袋和皂苷形成疏水作用。
综上可以得出,肾素和皂苷在6个活性口袋内都存在较强的疏水相互作用,并明确得出了每个活性口袋内形成疏水相互作用的关键残基氨基酸。
3 结语
对大豆皂苷I及其类似物大豆皂苷II、甘草皂苷和单葡萄糖醛酸甘草皂苷元与肾素进行分子对接,并对4种复合物分别采用分子动力学模拟和MM-PBSA方法计算结合自由能,计算结果和实验值测得的抑制剂抑制能力相接近。通过对结合能能量分解表明范德华力和静电力是形成能够形成复合物的主要驱动力,非极性溶剂化能对于复合物形成的促进作用较小。通过活性口袋氢键分析可以得出,S2、S3和S1是肾素和皂苷形成氢键作用重要的活性口袋,Ser230、Tyr231和Ser84等氨基酸残基是肾素和皂苷类抑制剂形成氢键作用的主要的氨基酸残基。范德华力主要来自于非极性氨基酸形成的疏水作用,通过Ligpiot软件以及分析活性口袋内的疏水作用得出,每个活性口袋是肾素和皂苷形成疏水作用的活性区域,Ala229、Asp38、Asp226、Gly228和Tyr83是肾素与皂苷类抑制剂结合过程中形成疏水作用的重要氨基酸残基。

表5 肾素活性口袋内与皂苷具有疏水作用的氨基酸残基Table 5 Amino acid residues inside renin activity pockets interaction with saponins by hydrophobic effect
[1]Gwathmey T M,Pendergrass K D,Reid S D,et al.Angiotensin-(1-7)-Angiotensin-Converting Enzyme 2 Attenuates Reactive Oxygen Species Formation to Angiotensin II Within the Cell Nucleus[J].Hypertension,2010,55:166-171.
[2]陈凯先,蒋华良,嵇汝运.计算机辅助药物设计:原理,方法及应用[M].上海:上海科学技术出版社,2000.
[3]Reinikainen T L,Ruohonen L,Nevanen T,et a1.Investigation of the function of mutated cellulose-binding domains of Trichoderma reesei cellobiohydrolase I[J].Proteins:Structure,Function and genetics,1992,14(4):475-482
[4]Schrodinger,Llc.The PyMOL Molecular Graphics System,Version 1.3r1.2010.
[5]王红寅,林东强,姚善,等.4-巯乙基吡啶配基与IgG相互作用的分子模拟研究[J].化学学报,68(16):1597-1602.
[6]Wallace A C,Thornton J M,Laskowski R A.LIGPLOT:a program to generate schematic diagrams of protein-ligand interactions[J].Protein Engineering,1995,8(2):127-134.
[7]Zhang Y,Pan D B,Shen Y L,et al.Understanding the molecular mechanism of the broad and potent neutralization of HIV-1 by antibody VRC01 from the perspective of molecular dynamics simulation and binding free energy calculations[J].Journal of Molecular Modeling,2012,18:4517-4527.
[8]Wang J,Morin P,Wang W.Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of Efavirenz by docking and MM-PBSA[J].American Chemical Society,2001,123:522l-5230.
[9]De Grandis V,Bizzarri A R,Cannistraro S.Docking study and free energy simulation of the complex between p53 DNA-binding domain and azurin[J].Journal of Molecular Recognition,2007,20:215-226.
[10]Takamatsu Y,Sugiyama A,Purqon A,et al.Binding free energy calculation and structural analysis for antigen-antibody complex[J].AIP Conference Proceedings,2006,832:566-569.
[11]Schrodinger,Llc.The Pymol molecular graphics system,Version 1.3r1.2010.
[12]Guang C E,Shang J G,Jiang B.Transport of traditional Chinese pimple milk-derived angiotensin-converting enzyme(ACE)inhibitor peptides across a Caco-2 cell monolayer and their molecular recognition with ACE[J].Journal of Food,Agriculture&Environment,2012,Vol.10(3&4):40-44.
[13]Takahashi S,Hori K,Hokari M,et al.Inhibition of human renin activity by saponins[J].Biomedical Research,2010,31(2):155-159.
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
中华养生保健(2020年2期)2020-11-16
心肺血管病杂志(2019年12期)2019-05-20
中学化学(2015年12期)2016-01-19
池州学院学报(2015年3期)2016-01-05
天津科技大学学报(2015年2期)2015-08-09
现代检验医学杂志(2015年5期)2015-02-06
原子与分子物理学报(2014年3期)2014-02-28
无机化学学报(2014年1期)2014-02-28

- 食品与生物技术学报的其它文章
- 复合改性改善大豆分离蛋白功能性质的研究
- 原生贡寮山药萃取物抗氧化成分及抗氧化活性的研究