
肼催化还原U(Ⅵ)制备U(Ⅳ)的研究进展
2014-12-25 07:47胡思思夏良树彭安国肖静水
核化学与放射化学 2014年1期
胡思思,夏良树,彭安国,肖静水,陈 伟
1.南华大学 化学化工学院,湖南 衡阳 421001;2.南华大学 核科学技术学院,湖南 衡阳 421001
核能是世界上继煤、石油和天然气等化石燃料之后的第三种主要能源,有望成为我国未来主要能源形式[1]。核能虽然具有储藏能容易获得、能量密度高、空间障碍较小及无空气污染等优势而快速发展[2],但是积累的大量乏燃料也成为一大问题。怎样处理这些放射性强、毒性大的乏燃料,一直存在着争议。处理技术路线主要有两种:一是美国所推崇的开环式或“一次通过”式核燃料循环;另一种则是英法等国坚持的闭合式燃料循环,即对乏燃料进行一些化学处理,从中回收大量的铀与少量的钚,并通过再循环加以充分利用[3]。天然铀资源的有限性使得实现核燃料的循环利用成为必然,其中乏燃料后处理是裂变核能可持续循环利用的关键环节。它的主要目的是,用化学方法将乏燃料中的U、Pu以及核裂变产物(FP)相互分离,以回收并净化U、Pu作为核燃料再利用,同时减少放射性废物的排放[4-5]。
目前乏燃料后处理工艺分干法和湿法两种。湿法包括沉淀法、溶剂萃取法、离子交换法等,干法包括氟化物挥发法、干式高温法等。至今国内外对生产堆乏燃料的处理流程大多数采用Purex循环流程,该流程的基本原理是,利用铀、钚和裂变产物的不同价态在有机萃取剂中有不同的分配系数,将它们一一分离[6]。在Purex流程中,采用磷酸三丁酯(TBP)作萃取剂,煤油作稀释剂,硝酸铀(Ⅳ)作Pu(Ⅳ)的还原剂,从硝酸溶液中萃取硝酸铀酰和Pu(Ⅳ)。而在该流程中以硝酸铀(Ⅳ)作Pu(Ⅳ)的还原剂具有还原反萃完全、不向系统引入杂质、反应速率较快及使不锈钢设备免受腐蚀等优点,已慢慢代替原来的氨基磺酸亚铁作为还原剂,广泛为各国轻水堆乏燃料后处理厂所采用。为了减少流程中铀的循环量和提高钚的浓缩倍数,一般后处理厂要求制备的U(Ⅳ)质量浓度大于150g/L,U(Ⅳ)占总铀浓度的百分含量大于75%。故在乏燃料后处理工艺中还原U(Ⅵ)制备U(Ⅳ)作为Pu(Ⅳ)的还原剂就显得尤为重要。
1 还原U(Ⅵ)制备U(Ⅳ)的早期研究
在乏燃料后处理方法的发展过程中,U(Ⅳ)溶液的制备一直广受关注,20世纪中期就有过相关的报道。1961年Katz等[7]已用沉淀法得到过硝酸铀(Ⅳ)溶液,这在当时相对来说是比较好的制备U(Ⅳ)溶液的方法。这种方法是通过向过量的2.5%硫酸铵中加入硝酸铀酰溶液得到硫酸铀(Ⅳ)沉淀,然后将硫酸铀(Ⅳ)分解得到UO2和硫磺,固体UO2可溶于60℃的硝酸中,而硫磺则以固体的形式被分离,控制条件还可以使整个硝酸铀溶液的形成过程无氧化反应发生。但是从以上过程中不难发现,其步骤比较繁琐,在实际运用中又因其他的一些影响因素而使要达到预想的结果比较困难,具有一定的局限性。而后Ondrejcin等[8]从还原性单质出发,选择一些活泼性适当的金属作为还原剂还原U(Ⅵ)制备U(Ⅳ)溶液,如在硫酸介质中用锌或者铅还原铀酰离子。锌和铅价格便宜,并且具有其独特的两性特征,使得他们能从氢氧化铀中分离出来,得到的沉淀物可以在以肼作为稳定剂的硝酸中溶解,但是此法得到的沉淀在其形成过程中易氧化,效果不够理想。
2 还原U(Ⅵ)制备U(Ⅳ)的近期研究
目前,还原U(Ⅵ)制备U(Ⅳ)的方法主要有以下3种:电化学法、光催化法和异相催化法。
2.1 电化学法
电解还原的原理是,依靠外加电压,使某些阳离子在阴极发生还原反应,同时另一些离子在阳极发生相应的氧化反应[9]。对于用电解法来制备U(Ⅳ)而言,电解池中除铀酰离子外,还有支持还原剂肼以及一定量的酸,高浓度酸更有利于U(Ⅳ)的形成[10]。一般阴极会发生铀酰离子转化为U(Ⅳ)离子的还原反应,而阳极则是肼离子的氧化反应。
国内外前期大多采用汞阴极电解还原制备U(Ⅳ),但由于汞的毒性较大,并且易污染环境,因此汞阴极电解还原法制备U(Ⅳ)的应用受到限制。而铂和钛有较好的耐腐蚀性能[11],因此何阿弟等[12]采用钛板作阴极,镀铂钛网作阳极,这也是国内目前多数人使用的电极材料,在硝酸体系中进行铀的电解还原[13],无论电解体系中有无隔膜存在,镀铂钛网都会受到一定程度的腐蚀[11],但这是无法避免的。在此基础上,为提高U(Ⅳ)的转化率,也有人使用过脉冲柱[9]、动态连续电解[14]、阳离子交换膜[15]等方法,而针对Arai等[16]使用玻璃碳纤维作阴极的封闭系统,存在的钯和银在阴极沉积堵塞以及电解产物破坏肼等问题,Nogami等[17]用碳感材料作为电极形成恒定电流,并且电解池与外界相通,就能很好的解决这一问题。
2.2 光催化法
光催化法具有反应系统简便,易于控制,便于维修及所用的还原剂无残渣等独特的优点,故光催化还原法制备硝酸铀(Ⅳ)得到国内外研究者的较大关注[18-24]。
光催化还原铀酰是指铀酰离子在光源的照射下,吸收一定能量成为激发态离子,激发态离子或与还原剂反应生成U(Ⅴ)[23],再歧化为U(Ⅵ)和U(Ⅳ);或者铀酰离子与溶液中的有机基质相结合[22],然后在照射下形成激发态络合物,猝灭后生成U(Ⅳ),通过哪种方式反应,这取决于不同还原剂的性能。
早期,Katz等[7]初步研究过光催化法,但没有使用保护气体,且还原剂选择不当,使得光催化结果并不好,因此没有进行深入的探讨研究。20世纪80年代,徐鸿桂等[24-26]利用氩气激光为光源,进行了乙醇、肼和甲酸体系中的一系列实验,讨论各影响因素对反应的影响,并认为甲酸体系能避免肼体系中铵离子和氮气产生。国外也有很多研究使用汞灯为光源,以草酸[22,27-29]、柠檬酸[30]、甲酸[22]和二烃硫化物[31]为还原剂进行光催化UO2+2还原,其中以研究草酸的居多,这样可以处理一定浓度的铀溶液。而近年来出现的以TiO2为催化剂、在其表面进行光催化的研究[32-34],多用于含铀废水处理。
2.3 异相催化法
异相催化[35]又称多相催化或接触催化,指催化剂和反应物存在于不同的相中,在催化剂界面上引起的催化反应。对于一个多相催化反应来说,其反应过程包括:扩散、吸附、反应和脱附等几个过程。反应中最重要的是使用固体催化剂,反应物为液相或气相,催化反应在两相间的界面上发生。正因如此,异相催化剂的分离比较简单,以物理方式(如过滤、离心等)就可以将催化剂与产物分离。可重复使用次数高和低分离成本是这类催化剂的优势。异相催化剂虽然方便分离,但也因为物理相不同,在机制上,只能催化吸附在其表面上的分子,一定要等生成物离去后,反应物才能再次附着(如图1)。故催化剂总表面积大小与分子扩散速率快慢,是影响异相触媒催化效率的重要因素,这对研究反应类型及反应速率变化意义重大。
异相催化法相对来说条件要求低、装置简便、更易于工业化生产,本文将对目前肼异相催化U(Ⅵ)制备U(Ⅳ)的研究进行介绍。在肼催化U(Ⅵ)制备U(Ⅳ)的研究过程中,就是以固体铂或钯为催化剂,以酸溶液为介质,采用有机还原剂,反应发生在催化剂的表面,即两相之间,因此肼催化U(Ⅵ)制备U(Ⅳ)的反应是典型的异相催化反应。
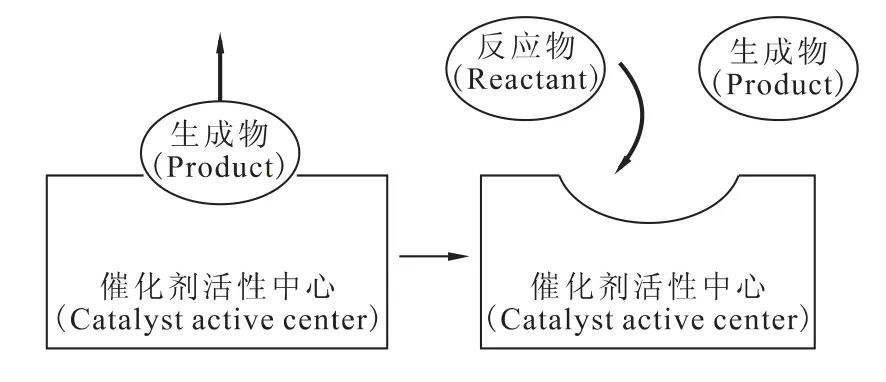
图1 催化剂结合反应物示意图Fig.1 Scheme of a catalyst binding the reactant
1)有机还原剂肼的选择
在肼催化U(Ⅵ)制备U(Ⅳ)的异相催化反应中,硝酸为强氧化性酸,且在高浓度中易产生化学性质更加活泼的亚硝酸,这就使得已由U(Ⅵ)还原的U(Ⅳ)又会被氧化为U(Ⅵ);而在非氧化性酸中,空气中的氧气在较高温度时也能氧化U(Ⅳ),因此保持U(Ⅳ)的稳定性尤为重要。
曾使用过的稳定剂主要有氨基磺酸[36]、邻氨基苯酸等胺类化合物、羟胺、肼[37-39]和尿素[39-40]等,其作用在于破坏硝酸中的亚硝酸[40]。
胺类稳定剂与HNO2反应式如下:
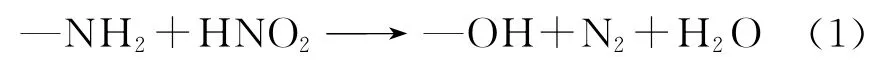
氨基磺酸、邻氨基苯酸与HNO2反应易引入磺酸基、苯甲酸基等外来杂质,这会增加废液量且影响废液的回收;另外氨基磺酸与亚硝酸反应时生成硫酸根,会对设备造成腐蚀。
羟胺、肼和尿素稳定剂具有较强的还原性,常作为稳定还原剂。王万春等[41]经研究发现,这3种稳定剂中,羟胺稳定U(Ⅳ)的效果最佳,尿素和肼对其稳定作用相近。但羟胺不稳定,室温吸收水汽和CO2后迅速分解,不利于储存。尿素因其价格便宜,虽然与亚硝酸反应速度不如胺类及肼,但也可以达到稳定U(Ⅳ)的作用。尿素与亚硝酸反应如下:
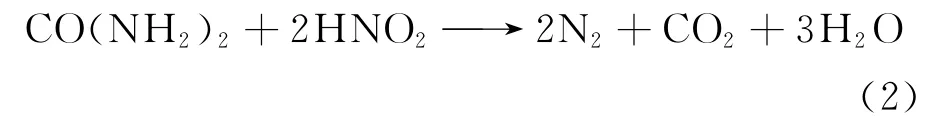
尿素易溶于水,因此易发生水解反应。而肼有较强的碱性及还原性,在高温(约100℃)下才分解为氮气、氨气和氢气。
单从还原性的角度来说,氢气[42]、甲酸和肼都可做还原剂。Boltoeva等[43]就使用过甲酸作还原剂,Swanson等[44]认为,对于后处理过程而言,肼作为还原剂优于氢作还原剂。
综上所述,肼是一种碱性的强还原剂,相对于Ondrejcin等[8]使用的金属单质还原剂来说,肼在U(Ⅳ)溶液的形成过程中的稳定作用是不可或缺的,而将稳定剂与还原剂结合为一种物质,毋庸置疑的给溶液处理提供了更大的便利,且肼易分解,不会给废液处理造成太大的困难,因此选取肼作为制备U(Ⅳ)的还原稳定剂是当今趋势。当然肼也有不如人意的地方,比如水合肼本身的毒性且易挥发、自身在酸溶液中也有可能发生氧化还原反应、高温下的不稳定以及催化反应后的成品中铵离子浓度较高等,而如何克服这些困难,找到适应的条件也是研究的一个重要方向。
2)肼催化还原U(Ⅵ)制备U(Ⅳ)的机理
肼催化还原U(Ⅵ)的反应机理随介质及催化剂的不同而有所差异。对于铂来说,在氧化性酸与非氧化性酸中的催化机制完全不一样。在硝酸介质中,还原剂肼可能会先被分解,然后再与铀酰离子反应,具体反应机理未见报道;而在非氧化性酸中,肼离子往往会先被催化剂吸附到其活性中心,从而得到中性肼,然后其活化态再进行还原反应。具体反应机理如下[45-46]:
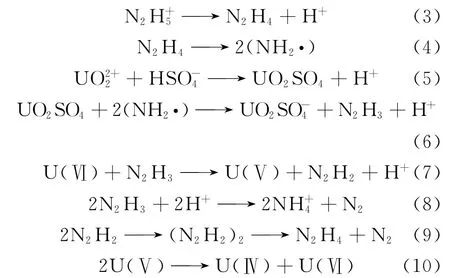
而对于金属钯来说,在硝酸中基本上不引发U(Ⅵ)还原,反而在硫酸介质中的催化效果较好[47],其中铀酰可能会被钯化学吸附后的肼自由基和氢离子2种物质还原:
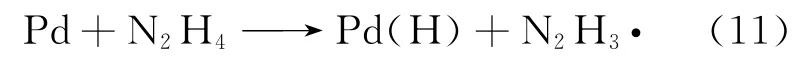
其还原过程分别为:
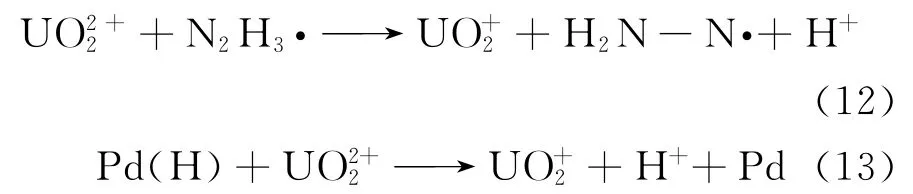
最后U(Ⅴ)再歧化为U(Ⅳ)与U(Ⅵ),如式(10)所示。
钯在硫酸中的催化反应主要分为2个过程[47]:吸附了氢离子的钯还原U(Ⅵ)的反应过程和被吸附后的肼自由基还原U(Ⅵ)的反应过程,具体过程示于图2。钯吸附肼的反应速率很快,而在还原铀酰的这2个反应过程中,钯吸附的氢与铀酰离子的反应比肼自由基还原铀酰的反应更慢一些,因此吸附的氢与铀酰离子的还原反应是决定反应速率的关键步骤,该反应为扩散控制(活化能约为19kJ/mol)。
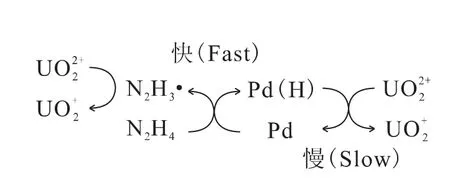
图2 钯在硫酸介质中的催化反应过程Fig.2 Catalytic process of Pd in sulfuric acid
对于同一催化剂,采用不同的介质(硝酸与硫酸)其反应机理也不相同,可能与硝酸的氧化性及硫酸易与反应产物络合有关;而对于相同的介质,采用不同的催化剂其机理也存在差异,可能是因为催化剂活性中心吸附能力的大小有差异,以及吸附后的产物与硝酸的反应性能不同所造成的。
3)肼催化还原U(Ⅵ)制备U(Ⅳ)在不同反应条件下的比较
在肼催化还原U(Ⅵ)制备U(Ⅳ)的异相催化还原反应中,一般以肼为还原剂,铂黑[44-46,48]或钯[47]为催化剂,在硝酸[44,48]、硫酸[45,47-48]或高氯酸[46,48]反应介质中进行。反应以氩气鼓泡来搅拌的居多,这样可以减少催化剂的损耗,少数实验用磁力搅拌,这种搅拌方式比较方便,易于达到。
(1)催化剂的预处理
以铂为催化剂时,一般都会对铂催化剂进行一些预处理。Swanson等[44]将铂置于氧化铝基质上,Abdunnabi等[45]将铂放在粒径为0.250~0.59mm的硅胶上,这样相比于直接使用铂黑粉末来说,能防止催化剂结块,以便得到分散性更好的催化剂,充分发挥其催化效能。而相对于铂来说,钯的稳定性稍差,需要进行一些还原性处理。处理钯催化剂的方法主要有以下两种[47]:一是将催化剂在350℃下焙烧2h,然后在400℃的高温下用肼还原2h;二是直接在400℃高温中用氢气还原2h。相比较而言,使用方法一时得到的催化剂活性较高,这可能是因为预热时已将多余的水分及部分杂质除去,使后面的还原过程进行的更彻底,这也体现了肼具有较好的还原效果。
(2)不同介质对肼催化还原U(Ⅵ)制备U(Ⅳ)反应的影响
目前肼催化还原U(Ⅵ)制备U(Ⅳ)大多在硝酸[44,48]、硫酸[45,47-48]或高氯酸[46,48]反应介质中进行,因硝酸具有其独特的较强的氧化性,故它与硫酸、高氯酸存在一定的差异。
①反应速率不同。由于其机理间的差异,导致反应物中肼对反应速率的影响也不一样。在硝酸介质中,U(Ⅳ)的生成会随肼的增加而增加,而硫酸溶液中U(Ⅳ)的量则不会随肼而变化,这与硫酸介质中催化剂吸附肼的过程有关,也是异相催化反应的特征,即催化剂结合肼的活性中心数是一定的,因此反应速率到达一定程度后不会随肼的增加而无限上升。
②催化剂的选择性不同。在非硝酸介质中,铂催化剂在载体上的分散性会影响催化剂的选择性,颗粒度的大小对肼的分解和铀的还原具有不同的影响;而在硝酸溶液中无论是肼分解反应还是铀还原的反应中,催化剂在一定尺寸范围内,活性都会随尺寸的加大而增加[43,48]。
③钯在不同介质对肼催化还原U(Ⅵ)制备U(Ⅳ)反应的影响。钯在硫酸介质中不会催化肼分解,因此对于肼催化还原U(Ⅵ)制备U(Ⅳ)的过程,在硫酸介质中U(Ⅵ)几乎完全还原转化为U(Ⅳ);而硝酸介质中只会引起肼的分解,且在该介质中钯催化剂还会有微量的溶解,这可能与硝酸的氧化性有关。另外,硝酸中的氢离子会促使肼分解的更快。钯的催化反应活性在硝酸、高氯酸、硫酸介质中依次增大,这主要是因为硫酸介质与反应产物形成了络合物。
④无论选择何种催化剂,也不论在何种介质中,肼催化还原U(Ⅵ)制备U(Ⅳ)的反应都有相似之处。反应介质酸的浓度与反应速率成反比。这是溶液中氢离子与肼离子在催化剂活性中心的吸附形成竞争的缘故,也与硝酸中氢离子促进肼分解有关,同时酸溶液改变了溶液中的离子强度对其也有一定的影响。在反应开始的短期时间内,反应速率对初始U(Ⅵ)浓度的反应级数为1。催化剂表面铂的活性与金属晶体平均尺寸成正比。反应活化能均为正,说明升温可以加快反应速率。
2.4 肼催化还原U(Ⅵ)制备U(Ⅳ)的反应热力、动力学
总的来说,在肼催化还原U(Ⅵ)制备U(Ⅳ)的反应过程中,铀酰初始浓度、肼浓度、温度和催化剂用量与反应速率成正比,催化剂的用量对反应转化率无影响。
在硫酸介质中,当铀浓度为0.05mol/L时,符合一级反应,对肼的反应级数为0.5,对酸反应级数为-0.42。硫酸的浓度范围最好在0.1~1mol/L,酸度太小易发生水解,酸度太大会有硫酸肼晶体析出[48]。在高氯酸中的反应动力学方程[48]为:
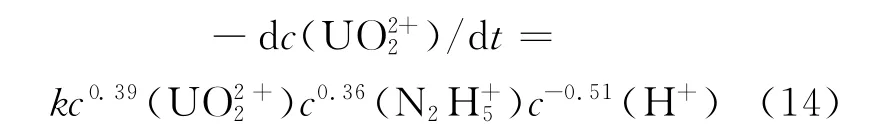
在硝酸介质中的动力学研究还未见报道。肼催化还原U(Ⅵ)制备U(Ⅳ)的反应为异相催化反应,因此也具有异相反应的一些独特性质,主要表现在以下两个方面:
(1)在动力学方面,反应级数是不确定的,随着初始U(Ⅵ)的浓度而变化,在痕量浓度下对铀一般为一级动力学,但在高浓度时又会不同,这与了解到的均相反应具有特定的反应级数完全不同,是异相催化反应的显著特征;
(2)在热力学方面,ln k-T-1的图像曲线是非线性关系,随着体系温度的升高,反应由动力学控制(Ea=30.1kJ/mol)转变为扩散控制(Ea=7.8kJ/mol);另外吸附肼的催化中心在肼浓度到一定程度后会达到饱和,这也充分体现了异相反应的特征。
3 研究展望
乏燃料后处理工艺中每种制备U(Ⅳ)的方法都具有一定的优越性,同时每种方法也存在一定的局限性。相比较而言,肼异相催化还原法具有还原反萃完全、不向系统引入杂质、反应速率较快及使不锈钢设备免受腐蚀等优势,具有较好的应用前景。前人的研究主要集中在工艺条件、反应机理、反应介质的选择、催化剂活性中心等的研究上,而对于反应过程的热力、动力学,特别是在硝酸介质中,研究分析的比较少,因此可以进行进一步的研究。
对于催化剂,其中毒现象讨论的并不多,只有Swanson[44]提到过,并说明在硝酸(而并不是硝酸肼)的冲洗下,可以使中毒催化剂复活,而对于昂贵的催化剂来说,若能解决中毒问题,使催化剂重复循环利用,那么对于大批量的反应来说无疑减少了大量的成本。
在催化剂的选择方面,可以结合经济性与反应性能综合考虑,寻找更为实用、环保、廉价的催化剂,如文中的钯催化剂;或者研究一些性能更好的双组分加成型催化剂[49],在保证较好催化活性的同时降低昂贵金属的使用量,这值得我们更加深入的探讨与研究。
[1]叶国安,张虎.核燃料后处理技术发展及其放射化学问题[J].化学进展,2011,23(7):1289-1294.
[2]侯媛媛.硝酸体系、高氮酸体系和有机相中U(Ⅳ)的稳定性研究[D].北京:中国原子能科学研究院,2004.
[3]任凤仪,周镇兴.国外核燃料后处理[M].北京:原子能出版社,2004.
[4]韦悦周.国外核燃料后处理化学分离技术的研究进展及考察[J].化学进展,2011,23(7):1272-1288.
[5]周贤玉.核燃料后处理工程[M].哈尔滨:哈尔滨工程大学出版社,2009.
[6]李民权.核工业生产概论[M].北京:原子能出版社,1995.
[7]Katz H.Some methods of preparing tetravalent uranium solutions[R].Washington D C:the Office of Technical Services,Department of Commerce,1961.
[8]Ondrejcin R S.Preparation of uranium(Ⅳ)nitrate solution[M].US:The United States Atomic Energy Commission,1962.
[9]邰德荣,仝继红,王欣昌,等.硝酸铀酰在电还原脉冲筛板中的电解还原[J].原子能科学技术,1992,26(3):1-12.
[10]Wei Y Z,Fang B,Arai T,et al.Electrochemical reduction of uranium(Ⅵ)in nitric acid-hydrazine solution on glassy carbon electrode[J].J Radioanal Nucl Chem,2004,262(2):409-415.
[11]丹尼尔·艾伦.钛在能源与工业中的应用[M].张祖光等,译.北京:机械工业出版社,1989.
[12]何阿弟,叶明吕,周祖铭,等.电解还原制备U(Ⅳ)过程中的阳极材料腐蚀研究[J].核化学与放射化学,1998,20(1):55-59.
[13]何阿弟,叶明吕,周祖铭,等.隔膜电解还原制备四价铀的研究[J].核技术,1997,20(7):413-417.
[14]何阿弟,叶明吕,周祖铭,等.动态连续电解还原制备四价 铀 的 研 究[J].核 技 术,1998,21(10):624-628.
[15]Smirnova N M,Glukhova L P,Glazkova I N,et al.Industrial operation of electrolyzers with cationite membranes for the reduction of uranium(Ⅵ)to uranium(Ⅳ)[J].Atomic Energy,1999,86(5):339-342.
[16]Arai T,Wei Y Z,Kumagai M.Proceedings of Global 2005[C].Tsukuba,Japan,October 9-13,2005.
[17]Nogami M,Hirose Y,Arai T,et al.Reduction of U(Ⅵ)and some fission products in HNO3media by galvanostatic electrolysis[J].J Alloys Compd,2008,451:358-360.
[18]Sidhu M S,Kohli K B,Bhatia P V K,et al.Photochemical reduction of uranylion with triethylamine[J].J Radioanal Nucl Chem,1994,187(5):375-383.
[19]Yusov A B,Shilov V P.Photochemistry of f-element ions[J].Russian Chemical Bulletin,2000,49(12):1925-1953.
[20]Bergamini P,Sostero S,Traverso O.f-element photochemistry[J].Fundamental and Technological Aspects of Organo-f-Element Chemistry,1985,155(1):361-385.
[21]付宏祥,吕功煊,李新勇,等.重金属离子的光催化还原研究进展[J].感光科学与光化学,1995,13(4):325-333.
[22]McCleskey T M,Foreman T M,Hallman E E,et al.Approaching zero discharge in uranium reprocessing:photochemical reduction of uranyl[J].Environ Sci Technol,2001,35(3):547-551.
[23]陆志人,陈兴华,袁顺庆,等.硝酸铀酰光化还原的研究[J].核化学与放射化学,1982,4(3):161-166.
[24]徐鸿桂,胡景忻,张先业,等.铀酸离子光化学研究Ⅰ:Ar+激光对UO2(NO3)2-C2H5OH-HNO3体系U(Ⅵ)的光化学还原[J].核化学与放射化学,1983,5(4):266-272.
[25]段云富,徐鸿桂,张先业,等.铀酸离子光化学研究Ⅱ:Ar+激光对肼体系U(Ⅵ)的光化学还原[J].核化学与放射化学,1984,6(3):125-130.
[26]张先业,胡景忻,周志宏,等.铀酸离子光化学研究Ⅲ:Ar+激光对UO2(NO3)2-HCOOH-HNO3体系U(Ⅵ)的光化学还原[J].核化学与放射化学,1985,7(3):141-146.
[27]Volman D H,Seed J R.The photochemistry of uranyl oxalate[J].J Am Chem Soc,1964,86(23):5095-5098.
[28]McBrady J J,Livingston R.The formation of tetravalent uranium during the uranyl-sensitized photochemical decomposition of oxalic acid[J].J Phys Chem,1946,50(3):176-190.
[29]Heidt L J,Tregay G W,Middleton Jr F A.Influence of pH upon the photolysis of the uranyl oxalate actinometer system[J].J Phys Chem,1970,74(9):1876-1882.
[30]Ohyoshi A,Ueno K.Studies on actinide elementⅥ:photochemical reduction of urany ion in citric acid solution[J].J Inorg Nucl Chem,1974,36:379-384.
[31]Sandhu S S,Kohli K B,Brar A S.Photochemical reduction of the uranyl ion with dialkyl sulfides[J].Inorg Chem,1984,23(22):3609-3612.
[32]Amadelli R,Maldotti A,Sostero S,et al.Photodeposition of uranium oxides onto TiO2,from aqueous uranyl solutions[J].J Chem Soc Faraday Trans,1991,87(19):3267-3273.
[33]Chen J,Ollis D F,Rulkens W H,et al.Photocatalyzed deposition and concentration of soluble uranium(Ⅵ)from TiO2suspensions[J].Colloids Surf A Physicochem Eng Asp,1999,151:339-349.
[34]Bonato M,Allen C C,Scott T B.Reduction of U(Ⅵ)to U(Ⅳ)on the surface of TiO2anatase nanotubes[J].Micro &Nano Letters,2008,3(2):57-61.
[35]傅献彩,沈文霞,姚天扬,等.物理化学(下册)[M].北京:高等教育出版社,2009:284.
[36]Jenkins E N,Stretion R J W.The use of uranium(Ⅳ)as a reagent in the aqueous processing of irradiated uranium[R].UK:United Kingdom Atomic Energy Authority,1959.
[37]Buckingham J S,Colvin C A,Goodall C A.The use of tetravalent uranium and hydrazine as partitioning agents in solvent extraction process for plutonium and uranium[R].Richland,American:General Electric Co.Hanford Atomic Products Operation,1959.
[38]Mckay H A C,Streeton R J W,Wain A G.Runs to study uranium(Ⅳ)as a reductant in uranium/plutonium separation[R].Harwell,United Kingdom:Atomic Energy Research Establishment,1963.
[39]Streeton R J W,Jenkins E N.The preparation,stabilization and analysis of uranium(Ⅳ)nitrate solutions[R].Harwell,United Kingdom:Atomic Energy Research Establishment,1962.
[40]周大凡,彭安,王承宪.硝酸铀(Ⅳ)稳定性的研究[J].原子能科学技术,1979,13(1):66-70.
[41]王万春,周祖铭,叶明吕.硝酸铀(Ⅳ)-磷酸三丁醋萃取体系中铀(Ⅳ)的稳定性[J].复旦大学学报,1997,36(1):47-52.
[42]Ananiev A V,Tananaev I G,Shilov V P.Heterogeneous catalytic redox reactions in the chemistry and technology of the nuclear fuel cycle[J].Russ Chem Rev,2005,74(11):1039-1059.
[43]Boltoeva M Y,Shilov V P,Anan’ev A V.Catalytic reduction of U(Ⅵ)with formic acid in acid solutions on palladium catalysts[J].Radiochem,2008,50(1):46-51.
[44]Swanson J L.Platinum catalyzed reduction of plutonium(Ⅳ)and uranium(Ⅵ)[R].US:Atomic Energy Commision,1971.
[45]Abdunnabi H M,Ananyev A V,Krot N N.Platinum catalyzed reduction of uranium(Ⅵ)with hydrazine in sulfuric acid media[J].J Radioanal Nucl Chem,Letters,1994,186(1):89-97.
[46]李斌,何辉,张秋月,等.高氯酸体系中肼为还原剂催化还原U(Ⅵ)的反应动力学[J].核化学与放射化学,2012,34(4):213-217.
[47]Boltoeva M Y,Trefilova A V,Anan’ev A V.Catalytic reduction of U(Ⅵ)with hydrazine on palladium catalysts in acid solutions[J].Radiokhimiya,2008,50(1):34-40.
[48]Boltoeva M Y,Shilov V P,Anan’ev A V.Reactivity of platinum nanoaggregates in catalytic reduction of U(Ⅵ)with hydrazine in acid solutions[J].Radiokhimiya,2007,49(6):529-531.
[49]黄旭,韩青霞,李娟,等.铂-钯/活性炭催化剂的制备及其在双组分加成型液体硅胶中的应用[J].云南大学学报,2012,34(2):218-223.
猜你喜欢
能源工程(2021年1期)2021-04-13
化学与生物工程(2021年2期)2021-02-25
中学生数理化·高一版(2020年11期)2020-12-14
无机材料学报(2020年2期)2020-03-08
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
中学化学(2019年2期)2019-07-08
分析化学(2019年3期)2019-03-30
中国资源综合利用(2016年6期)2016-01-22
核科学与工程(2015年2期)2015-09-26
